
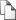
Преждевременное старение и радиочувствительность клеток у больных атаксией-телеангиэктазией
На правах рукописи
ПОЛУБОТКО Евгения Анатольевна ПРЕЖДЕВРЕМЕННОЕ СТАРЕНИЕ И РАДИОЧУВСТВИТЕЛЬНОСТЬ КЛЕТОК У БОЛЬНЫХ АТАКСИЕЙ-ТЕЛЕАНГИЭКТАЗИЕЙ 03.00.25 Гистология, цитология, клеточная биология
АВТОРЕФЕРАТ
диссертации на соискание ученой степени кандидата биологических наук
САНКТ-ПЕТЕРБУРГ 2009
Работа выполнена в Лаборатории радиационной цитологии Учреждения Российской академии наук Института цитологии РАН, Санкт Петербург
Научный консультант: кандидат биологических наук, доцент Спивак Ирина Михайловна Институт цитологии РАН, Санкт-Петербург
Официальные оппоненты: кандидат биологических наук Поспелова Татьяна Викторовна Институт цитологии РАН, Санкт-Петербург доктор биологических наук, профессор Воробцова Ирина Евгеньевна Российский научный центр радиологии и хирургических технологий Федерального агентства по высокотехнологической медицине
Ведущая организация: НИИ онкологии им. Н.Н.Петрова Росмедтехнологии
Защита состоится “ “октября 2009 года в ч на заседании Диссертационного совета Д.002.230.01 при Институте цитологии РАН по адресу: 194064, Санкт-Петербург, Тихорецкий пр., д.4.
Факс: +7(812)297 Сайт: www.cytspb.rssi.ru
С диссертацией можно ознакомиться в библиотеке Института цитологии РАН Реферат разослан “ ” сентября 2009 г.
Ученый секретарь диссертационного совета кандидат биологических наук Каминская Е.В.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы Функциональный анализ генов, вовлеченных в процессы репарации ДНК, является основой для понимания природы генетической нестабильности и повышенной радиочувствительности клеток у человека, приводящих к опухолевой трансформации, раку и старению (Sancar et al., 2004;
Thaker, Zdzienicka, 2005). Важный прогресс в этой области в течение последнего десятилетия, за время которого было клонировано и секвенировано большинство репарационных генов и подробно охарактеризованы кодируемые ими белки (Hoeijmakers, 2001;
Hanawalt, 2001;
Iliakis et al., 2003), значительно расширил наши представления о механизмах, позволяющих всем живым организмам сохранять собственную генетическую целостность после повреждающих воздействий. Важнейшие достижения в этой области были связаны с описанием редких наследственных заболеваний человека, возникающих при наличии мутаций в генах, вовлеченных в процессы репарации ДНК (Спивак, 1999;
Christmann et al., 2003). Эти заболевания являются естественной моделью для изучения процессов репарации ДНК и канцерогенеза, причем одновременно в семьях больных прослеживается предрасположенность к повышенной частоте возникновения раковых заболеваний, связанная с гетерозиготным носительством мутаций в тех же генах (Nakayama, 2002;
Pulverer, 2003). Клеточные линии и штаммы, полученные от таких больных, являются моделью для изучения таких процессов, как репарация, репликация и рекомбинация ДНК, старение и канцерогенез (Михельсон, 1996;
Наседкина и др., 1997;
Спивак и др., 1997;
Спивак, 1999;
Barenfeld et al., 1998;
Mikhelson, 2001).
Изучение репарации ДНК шло параллельно с исследованиями действия различных повреждающих агентов на молекулярном уровне. Особое место среди повреждающих воздействий занимают ионизирующая радиация и химические мутагены, действие которых приводит к сходным последствиям, в первую очередь—возникновению двунитевых разрывов ДНК (Kolman et al., 1997;
Chovanec et al., 1998;
Huang et al., 2003). Появление в ДНК двунитевых разрывов (DSBs) запускает каскад внутриклеточных реакций, опосредуемых различными сигнальными путями, и приводит к развитию глобального клеточного ответа на повреждение, включающего активацию специфических чекпойнтов (мест сшивок), возникающих в ответ на повреждение ДНК. Следствием этого является остановка клеточного цикла, усиление репарационных процессов и изменение конформации хроматина (Belayev et al., 1996;
Fei, El-Deiry, 2003;
Iliakis et al., 2003;
Zhou, Bartek, 2004). Ключевым белком глобального клеточного ответа принято в настоящее время считать протеинкиназу АТМ (ataxia-telangiectasia mutated), способную к автофосфорилированию и активации в ответ на возникновение конформационных изменений хроматина после появления DSBs и быстрому последующему фосфорилированию различных белков-мишеней, среди которых Р53, CHK2, BRCA1, NBS1, H2AX и многие другие (Kim et al., 2002;
Westphal et al., 1997;
Shiloh, 2003;
Lavin, 2008).
При наличии мутаций в обеих аллелях гена atm развивается тяжелое заболевание со сложным плейотропным фенотипом—атаксия-телеангиэктазия (АТ), или синдром Луи-Бара, характеризующееся множественными нейрологическими аномалиями, высокой предрасположенностью к опухолевым заболеваниям, ускоренным старением и крайней чувствительностью к действию ионизирующей радиации (Спивак, 1999;
Landsdorp, 2000;
Shiloh, 2003;
Lavin, 2008). При отсутствии АТМ ее функции частично берет на себя ATR (ataxia telangiectasia related), сходная с ней по структуре, но хуже изученная (Abraham, 2001). Ген АТМ находится на 11 хромосоме – 11q23, имеет размер примерно т.п.н. и содержит 66 экзонов (Uziel et al., 1996;
Platzer et al., 1997). Хотя к настоящему времени он полностью секвенирован, оказалось, что каждая детально исследованная АТ-семья несет свою собственную мутацию, что требует полного сиквенса исследуемого гена и существенно осложняет диагностику (Sandoval et al., 1999).
Несмотря на имеющиеся к настоящему времени многочисленные данные о роли белка АТМ в клеточном ответе на радиационные повреждения, остается недостаточно ясной картина в каждом отдельном случае, с которым сталкиваются клиницисты и ученые. К тому же гетерозиготное носительство этого заболевания гораздо шире, чем можно предположить, исходя из его частоты, и составляет по разным данным от 2 до 10 % в различных популяциях. Носители, как правило, имеют предрасположенность к развитию ранних опухолей различной этиологии.
Действие небольших случайных доз ионизирующей радиации или мутагенов может служить в таких случаях триггером для развития опухолевого процесса.
Поиск быстрого метода определения потенциального носительства мутаций в гене АТМ и других, отвечающих за процессы репарации ДНК в клетке, является, таким образом, одной из важных научно-практических задач радиобиологии и радиационной гигиены.
К настоящему времени накоплены многочисленные экспериментальные данные, указывающие на то, что для репарации DSBs необходимо ремоделирование хроматина, происходящее при строго ассоциированном с образованием DSB специфическом фосфорилировании С-концевого серина- вариантного гистона Н2АХ, получившего название -Н2АХ (Ward et al., 2001;
Goodarzi et al., 2008;
Kuo, Yang, 2008). Мегабазные участки хроматина, покрывающие DSB, можно легко визуализировать с помощью специфических антител на фосфорилированный короткий пептид, соответствующий С-концу Н2АХ. Эти фокусы могут служить маркерами DSBs и способствовать изучению их индукции и процессинга (Karlson, Stenerlow, 2004;
Tanaka et al., 2006). В клетках высших эукариот гистон Н2АХ фосфорилируется протеинкиназой ATM в первые же минуты после образования DSB, и инактивация АТМ приводит к нарушению этого фосфорилирования.
Еще одной прямой мишенью АТМ является белок Р53, фосфорилируемый ею по серину в 15 положении. Это фосфорилирование приводит к высвобождению Р53 из комплекса с MDM2 (который АТМ также фосфорилирует по серину в положении 395) и его стабилизации и накоплению в ядре. Благодаря стабилизации белка Р53 и его последующей, относительно более медленной, работе как фактора транскрипции, включающего синтез соответствующих белков, например белка Р21, в клетке генерируется длительная остановка в фазе G1, опосредуемая инактивацией комплекса циклин Е/CDK2 (Sancar et al., 2004).
Одновременно происходит взаимодействие белка Р53 с белком 53ВР1, его перемещение и формирование фокусов репарации. Другой важнейший фактор транскрипции, играющий ключевую роль в контроле клеточного цикла—белок E2F1, он также фосфорилируется и стабилизируется АТМ-зависимым способом (Lin et al., 2001). В то же время давно показана и многократно подтверждена независимыми исследователями радиорезистентность клеточного цикла в клетках больных АТ (Jeggo et al., 1998). Спорным остается вопрос о том, как изменятся параметры клеточного цикла в клетках гетерозиготных носителей АТ и будет ли другим характер формирования в них чекпойнтов в ответ на повреждение ДНК.
Повреждения ДНК, безусловно, вовлечены в появление и развитие такой возрастной патологии, как опухолеобразование, а гетерозиготное носительство мутаций в генах репарации ДНК и глобального клеточного ответа, вероятно, является причиной раннего возникновения опухолей различной локализации. Эти же мутации могут приводить и к более раннему появлению других заболеваний, характерных для пациентов старшего возраста, например, диабета и сердечно сосудистой патологии (Sano et al., 2007), то есть ускоренному старению. Это соответствует современным представлениям о природе возникновения болезней старения и их индивидуальной генетической детерминированности (Анисимов, 2003).
Таким образом, сейчас возникла ситуация, когда для прикладной медицины крайне важно иметь возможность быстро и достоверно оценить повышенную радиочувствительность каждого конкретного пациента на клеточном уровне. Такая оценка может быть основой как для показаний к диспансеризации, так и к выработке соответствующих медицинских рекомендаций для диагностики и профилактики патологий, в первую очередь, ранних опухолей, у лиц группы риска. Разработка методов быстрого определения степени радиочувствительности и ускоренного старения на клеточном уровне (вне зависимости от генетического дефекта) крайне необходима для пациентов, которым рекомендован курс радиотерапии и для лиц, связанных с риском радиационного или химического поражения в силу своей профессиональной деятельности Цель и задачи работы Целью данной работы являлось исследование преждевременного старения и радиочувствительности на клеточном уровне в семьях больных атаксией-телеангиэктазией (АТ).
Для достижения этой цели были сформулированы следующие задачи:
1. Изучить репаративный потенциал клеток больных АТ и их кровных родственников методом электрофореза индивидуальных клеток (методом “ДНК-комет”).
2. Описать маркеры ускоренного старения в клетках больных АТ и их кровных родственников.
3 Изучить особенности клеточного цикла в ответ на действие рентгеновского облучения в клетках больных АТ и их кровных родственников.
4. Сравнить уровень преждевременного старения и радиочувствительности на клеточном уровне в различных семьях, в которых была диагностирована АТ.
Научная новизна работы 1. Впервые на примере двух семей доказано существование связи между признаками радиочувствительности и признаками ускоренного старения на клеточном уровне при атаксии-телеангиэктазии.
2. Впервые предложен быстрый адекватный метод дискриминации гетерозиготного носительства в семьях, в которых была диагностирована АТ.
Теоретическая и практическая значимость работы Показано, что при клеточной радиочувствительности различной этиологии одновременно выявляются маркеры ускоренного старения. На примере двух семей АТ доказана связь радиочувствительности и проявления признаков ускоренного старения на клеточном уровне. На основе полученных результатов выявлены молекулярные механизмы, лежащие в основе радиоиндуцированного старения.
Показана эффективность использования клеточных культур для дискриминации гетерозиготного носительства АТ. Выявление гетерозиготного носительства АТ в семьях с выработкой последующих клинических рекомендаций будет способствовать улучшению качества жизни пациентов за счет оценки рисков, своевременного и адекватного назначения терапии сопутствующих заболеваний.
Положения, выносимые на защиту 1. Атаксия-телеангиэктазия является не только синдромом повышенной радиочувствительности, но и синдромом преждевременного старения на клеточном уровне.
мутации гена atm демонстрируют черты 2. Гетерозиготные носители преждевременного старения и повышенной радиочувствительности на клеточном уровне.
3. Гетерозиготное носительство мутации в гене atm может быть выявлено как по повышенной радиочувствительности клеток, так и по наличию у носителя маркеров старения на клеточном уровне.
4. Повышенная радиочувствительность на клеточном уровне сопряжена с риском развития опухолевых заболеваний.
Апробация диссертации По теме диссертации опубликовано 6 печатных работ. Материалы диссертации были представлены на III Международной конференции молодых ученых (Харьков, 2008), конференции “Фундаментальные науки - медицине” (Москва, 2008, 2007гг), а также на научных семинарах Лаборатории радиационной цитологии Учреждения Российской академии наук Института цитологии РАН.
Финансовая поддержка работы Работа выполнена при финансовой поддержке программы Президиума РАН “Биологические науки - медицине” 2006-2009гг.
Структура и объем диссертации Диссертация изложена на страницах машинописного текста, содержит гистограмм, таблиц, иллюстрирована рисунками. Она состоит из введения, обзора литературы, материалов и методов, результатов исследования, обсуждения результатов, выводов и списка литературы. Список использованной литературы включает источников.
МАТЕРИАЛЫ И МЕТОДЫ Клеточные штаммы и их культивирование В работе были использованы клеточные культуры диплоидных фибробластов, полученные от больных АТ, гетерозиготных носителей мутации в гене atm, процедура проведена в Лаборатории радиационной цитологии Института цитологии РАН в ходе стандартного протокола из эксплантата кожи предплечья.
Штаммы диплоидных фибробластов, полученные от членов первой семьи (АТ6) – девушки 16 лет с АТ и ее фенотипически здоровых матери (M) и отца (F), были обозначены соответственно: AT6SP, AT(M)6SP и AT(F)6SP.
Штаммы диплоидных фибробластов, полученные от членов второй семьи (АТ8) – девочки 11 лет с АТ, ее фенотипически здоровых матери (M) и отца (F) были обозначены соответственно: AT8SP, AT(M)8SP и AT(F)8SP. Диплоидные фибробласты, полученные от больной с атаксией, возникшей в результате опухоли мозжечка, были обозначены AT7SP. В качестве контроля были использованы диплоидные фибробласты крайней плоти мальчика 11 лет—VH (Nygren et al., 1994;
Kolman et al., 1997), любезно предоставленные проф. Адой Кольман (A. Kolman, Стокгольмский университет, Швеция).
Клетки культивировали в питательной среде DMEM (Gibco, США) с добавлением 12 % фетальной сыворотки телят (Hyclone, США) и антибиотиков (100 ед./мл пенициллина,100 мкг/мл стрептомицина, Sigma, США). Клетки выращивали в пластиковых флаконах, на чашках Петри (Nunclon, США), а также на предметных стеклах, помещенных в чашку Петри, в CО2-инкубаторе при 37С, 5-7% СО2 в атмосфере повышенной влажности.
Для выяснения степени радиочувствительности фибробласты облучали на рентгеновской установке РУМ-17 в дозе 2 Гр.
Иммунофлуоресцентный анализ Клетки выращивали на поверхности предметных стекол, помещенных в чашки Петри. После достижения клетками субконфлюэнтного состояния их подвергали рентгеновскому облучению и инкубировали в течение различных промежутков времени, в качестве контроля использовали интактные образцы.
Отмытые от культуральной среды раствором PBS (Биолот, Россия) клетки фиксировали 10 мин в 3.7 %-ном растворе формальдегида в PBS на льду, затем продолжали фиксацию в течение 24 ч в 70 %-ном этаноле и промывали раствором PBS в течение 30 мин. Для перфорации цитоплазматической мембраны препараты инкубировали 5 мин в 3 %-ном растворе Тритон X- (Хеликон, Россия) на PBS. Для блокирования мест неспецифического связывания препараты инкубировали от 30 мин до 1 сут в 1 %-ном растворе BSA (Sigma, США) на PBS, затем промывали.
В качестве первых антител использовали IgG кролика против: белка HP1 (Abcam, Англия), фосфорилированного по серину в 139-м положении гистона H2AX, (Cell Signaling, США), белка 53BP1 (Abcam, Англия), моноклоноальные Waf антитела к белку p21 (Abcam, Англия), в разведении 1:150. В качестве вторых антител использовали козьи антикроличьи IgG, козьи антимышиные IgG, коньюгированные с флуоресцеина изотиоцианатом (FITC), в разведении 1:150 (Sigma-Aldrich Co., США). Антитела разводили в PBS с 0.1 %-ным BSA (Sigma, США). После инкубации с антителами стекла промывали по 30 мин в 0.1 %-ном растворе Tween-20 (Хеликон, Россия) в PBS. В качестве контроля специфичности иммунной реакции использовали препараты клеток, инкубированные только со вторыми антителами. После окрашивания препараты помещали в раствор пропилгаллата в 90 %-ном глицерине для предотвращения «выгорания» флуоресцентной метки. Для анализа воспроизводимости полученных результатов все тесты проводили в 2–3 повторах.
Метод “ДНК- комет” Метод включал в себя следующие этапы: приготовление гель-слайда, лизис клеток, щелочную инкубацию, электрофорез, нейтрализацию, окраску слайдов, анализ и обработку данных. Все этапы (кроме последнего), проводились в условиях минимального попадания света, при 4 С. Для получения гель-слайдов использовали предметные стекла, предварительно покрытые 1 %-ным водным раствором агарозы (Биолот, Россия). Далее 50 мкл клеточной суспензии смешивали с 150 мкл агарозы с низкой точкой плавления (Аgarosе-рrер. Ultrа 1оw-gelling and melting point LKB, Швеция) (1 %-ный раствор в РВS), имеющей температуру 37 С, и быстро наносили на подготовленное предметное стекло примерно по 100 мкл на каждый слайд, сверху накрывали покровным стеклом и оставляли на 5 мин при 4 С. Затем снимали покровное стекло и приступали к этапу лизиса. Непосредственно перед процедурой лизиса к лизирующему буферу (2.5 мM NaС1, 100мМ Nа2ЕDТА, 10мМ Тris рН10) добавляли детергент Тритон Х-100 (Хеликон, Россия) до конечной концентрации 1 %. Лизис проводился в течение 1 ч при С. Затем слайды подсушивали и подвергали дальнейшей процедуре щелочной обработки. Для щелочной инкубации и электрофореза использовали один и тот же буфер (1 мМ Nа2ЕDТА, 300 мМ NаОН, рН 13) и, следовательно, первая процедура проводилась сразу в электрофорезном танке для избежания лишних манипуляций со слайдами, которые могли бы привести к потере геля с поверхности предметного стекла. Длительность щелочной инкубации составляла 40 мин при температуре 4 С. Электрофорез проводили в течение мин (4 С, напряжение 5 В/см). После извлечения слайдов избыток электрофоретического буфера удаляли фильтровальной бумагой, слайды подсушивали 5-7 мин и нейтрализовали 3-кратной промывкой в 0.4 М Тris рН 7,5 по 5 мин. Для окраски слайдов использовали SYBR® Green I (Sigma, США) в 90%-ном глицерине (Биолот, Россия) в разведении 1.5 мкл на 10 мл буфера TE (Tris-EDTA, pH 7.5)(Sigma, США). Затем слайды помещали во влажную камеру и оставляли в холодильнике при 4 С до момента визуального анализа.
Микроскопия и анализ изображений После иммунофлуоресцентного окрашивания препараты анализировали с помощью лазерного сканирующего флуоресцентного микроскопа Zeiss LSM PASCAL. Для визуализации флуорофоров были использованы аргоновый ( нм) и гелиево-неоновый (543 нм) лазеры. Для получения изображений использовали сканирующий модуль микроскопа, управляемый с помощью компьютера и соответствующего программного обеспечения LSM 5 PASCAL.
Для анализа полученных изображений (определение уровня интенсивности флуоресценции) использовали программы LSM 5 PASCAL и «WCIF ImageJ 1.37m» (National Institute of Health, Maryland, США).
После электрофореза отдельно взятых клеток визуализацию “комет” проводили на флуоресцентном микроскопе Zeiss LSM 5 PASCAL (источник света-ртутная лампа, 250 ватт) длина волны возбуждающего света задавалась фильтром 516-560 нм, барьерный фильтр 590 нм, увеличение 40 ). Ввиду отсутствия специально адаптированной для метода компьютерной программы оценку “комет” проводили визуально. Для характеристики каждой линии проводили по три эксперимента, в ходе которых оценивали содержание клеток каждого типа: клетки без повреждений ДНК, клетки с некоторыми повреждениями ДНК, клетки со значительными повреждениями ДНК. Далее высчитывали площадь “комет” (S) по формуле S=N0+N1*0.5+N2*1, где N количество клеток, относящихся к типу a, N2 – количество клеток, относящихся к типу b, N2 – количество клеток, относящихся к типу c (рис. 1).
a b c Рис.1. Гель-электрофорез первичных фибробластов человека после -облучения в дозе 2Гр (увеличение 40, окраска SYBR Green I).
a- клетки без повреждений ДНК, b - клетки с некоторыми повреждениями ДНК, c - клетки со значительными повреждениями ДНК.
Метод проточной цитометрии В опыт брали все клетки с чашки Петри, ресуспендировали в PBS, добавляли РНКазу (0.25 мг/мл), PI ( пропидий йодид) – 50 мкг/мл и сапонин – 200 мкг/мл или 0.1% Тритон. Анализ производили на цитометре Odam (Brucker, Франция).
Обработка результатов Обработку результатов проводили с помощью программы MS Excel общепринятыми для медико-биологических исследований методами: расчет средней арифметической величины, среднего квадратичного отклонения, ошибки репрезентативности для каждого параметра в исследуемых группах клеток, сравнение средних значений по критерию Стьюдента с определением достоверности различий.
РЕЗУЛЬТАТЫ Определение гетерохроматинового маркера НР1-.
Окраска непрямым иммунофлуоресцентным методом с использованием антител к маркеру старения белку НР1- выявила существенные различия между интактными клетками всех исследованных штаммов. В табл.1 приведены значения интенсивности флуоресценции ядер фибробластов кожи, полученных от членов двух изученных семей. Интенсивность флуоресценции почти в два раза снижена в ядрах клеток больных АТ (AT6SP и AT8SP) по сравнению с ядрами клеток здорового донора, а в клетках их кровных родственников – гетерозиготных носителей заболевания – это снижение также достоверно, хотя и менее выражено. Следует отметить тот факт, что фибробласты больной с атаксией, возникшей в результате опухоли мозжечка (клетки AT7SP), обнаруживают более низкую интенсивность флуоресценции по сравнению с atm. Вероятно, данная клетками гетерозиготных носителей мутации гена больная имеет наследственную радиочувствительность на клеточном уровне.
Название штамма клеток Интенсивность флуоресценции, Хср ± Sx VH10 170.71 ± 1. AT8SP 103.72 ± 5. AT(F)8SP 142.44 ± 3. AT(M)8SP 149.54 ±1. AT6SP 93.01 ± 3. AT(F)6SP 134.45 ± 4. AT(M)6SP 142.52 ± 2. AT7SP 124.13 ± 2. Табл.1. Интенсивность флуоресценции белка HP1- в диплоидных фибробластах человека Статистически достоверные отличия от контроля при p0.001, где n-100 клеток.
Количество фосфорилированной формы гистона Н2АХ определяли иммунофлуоресцентным методом как в интактных клетках, так и в клетках всех исследованных штаммов после рентгеновского облучения. Интенсивность флуоресценции была измерена так же, как и в случае с белком НР1-. Показано, что количество фосфорилированной формы гистона -H2AX в ядрах интактных клеток больных с АТ превышает таковое в ядрах клеток здорового донора, а для atm гетерозиготных носителей мутантного характерно промежуточное состояние. Обнаружены различия в клетках в отношении активности репарационных процессов. Через 24 ч после облучения в клетках больных с АТ происходит восстановление лишь половины повреждений ДНК (рис.2, а, кривая 4;
рис.2, б, кривая 4), в то время как в клетках здорового донора этот процесс практически завершается (рис.2, а, кривая 1;
рис.2, б, кривая 1). В клетках гетерозиготных носителей мутантного гена atm репарация проходит быстрее, чем у больных, но динамика этого процесса существенно отличается от таковой в клетках здорового донора (рис.2, а, кривые 2, 3;
рис.2, б, кривые 2, 3).
Количественные изменения фосфорилированной формы гистона -H2AX в ядрах клеток больных с атаксией после облучения представлены на рис. 4, а.
а Флуоресценция, усл. ед.
0 б Флуоресценция, усл. ед.
40 20 инт 30 мин 2.5 ч 24 ч Рис. 2. Интенсивность флуоресценции фосфорилированной формы гистона - H2AX в ядрах фибробластов до и после рентгеновского облучения.
а – штаммы, полученные от членов семьи АТ6;
б – штаммы, полученные от членов семьи АТ8;
1 - VH10, 2 – AT6(F)SP, AT8(F)SP, 3 - AT6(M)SP, AT8(M)SP, 4 – AT6SP, AT8SP.
Определение количества белка 53ВР1.
Еще одним традиционным маркером репарационных процессов является белок 53ВР1 (Iwabuchi et al., 2008;
Lee et al., 2008). Обычно при окрашивании антителами к белку 53ВР1 в интактных клетках наблюдается его диффузное расположение, а при повреждении ДНК белок концентрируется в зоне репарации, образуя фокусы.Фокусы белка 53BP1 в ответ на повреждение ДНК обычно совпадают с фокусами фосфорилированного гистона -Н2АХ, так как они часто колокализуются в зоне двунитевого разрыва ДНК. В интактных клетках, полученных от больных с АТ (штаммы AT6SP и AT8SP), фокусы белка 53BP1 наблюдаются в 16 % и 18 клеток соответственно, в то время как в контрольной линии VH10 таких клеток всего 6. На рис. 3, а и б приведены графики, описывающие динамику формирования и элиминации фокусов белка 53ВР1 в штаммах фибробластов, полученных от членов обеих семей, а на рис. 4, б – от больных с атаксией.
Известно, что белок 53ВР1 связывается с фосфорилированным белком р53, накапливаясь в местах повреждения ДНК. Так как фосфорилирование белка р53 в ответ на повреждение ДНК в клетке осуществляет протеинкиназа АТМ, то повышение количества фокусов белка 53ВР1 в клетках, полученных от больных с АТ задерживается по сравнению с клетками здорового донора (рис.3, а, кривая 4;
рис.3, б, кривая 4). Через 2.5 ч после облучения (в то время как в клетках здорового донора уже начинается процесс элиминации фокусов белка 53BP1) их количество несколько возрастает, но не достигает уровня репарационного ответа, как в клетках здорового донора, и сохраняется на стабильно высоком уровне через 24 ч после облучения. В это время в клетках здорового донора подавляющее число фокусов уже элиминируется (рис.3, а, кривая 1;
б, кривая 1). В клетках гетерозиготных носителей мутантного гена данного заболевания уровень репарационного ответа почти совпадает с контрольным уровнем здорового донора, но количество фокусов, сохраняющихся через 24 ч, существенно выше, чем в контроле (рис.3, а, кривые 2 и 3;
рис.3, б, кривые 2 и 3). Неспособность полностью элиминировать фокусы белка 53ВР1 свидетельствует о персистировании в этих клетках повреждений ДНК и сопряженных с ними изменений конформации хроматина. Изменения количества белка в штамме клеток AT7SP, полученных от больной с атаксией, возникшей в результате опухоли мозжечка, носят промежуточный характер, сходный с таковым в штаммах, полученных от гетерозиготных носителей мутантного гена atm (рис.4, б, кривая 2).
а Доля клеток, % 40 б Доля клеток, % 60 инт 30 мин 2.5 ч 24 ч Рис. 3. Формирование фокусов 53ВР1 в ядрах фибробластов до и после рентгеновского облучения.
а – штаммы фибробластов, полученные от членов семьи АТ6;
б – штаммы фибробластов, полученные от членов семьи АТ8;
1 - VH10, 2 – AT6(F)SP, AT8(F)SP, 3 AT6(M)SP, AT8(M)SP, 4 – AT6SP, AT8SP.
а Флуоресценция, усл. ед.
0 б Доля клеток, % 100 40 4 20 инт 30 мин 2.5 ч 24 ч Рис. 4. Интенсивность флуоресценции фосфорилированной формы гистона - H2AX (а) и формирование фокусов 53ВР1 (б) в ядрах фибробластов больных с атаксией до и после рентгеновского облучения.
1 - штамм VH10, 2 - штамм AT7SP, 3 - штамм AT6SP, 4 – штамм AT8SP.
p21Waf1/Cip1 - ингибитора Определение циклинзависимых киназ иммунофлуоресцентным методом в штаммах фибробластов, полученных от членов семьи AT8 после рентгеновского облучения, показало, что данный белок появляется в клетках больных АТ в ответ на повреждение ДНК с существенной задержкой. В клетках гетерозиготных носителей мутантного гена atm образование белка p21Waf1/Cip1 также запаздывает в отличие от контроля (Рис.5), но крайне незначительно.
инт. кл. 30 мин 2.5 ч 24 ч AT8SP AT(F)8SP AT(M)8SP VH Waf1/Cip Рис. 5. Интенсивность флуоресценции p21 в ядрах фибробластов до и после рентгеновского облучения Рис. 6. Иммунофлуоресцентное окрашивание клеток HP1-, где а – VH10, b – AT8SP, c – AT8(F)SP, d – AT7SP (увеличение 40).
a a b b c c d d Рис. 7. Иммунофлуоресцентное окрашивание клеток -H2AX, где а – VH10, b – AT8SP, c – AT8(F)SP (увеличение 40).
a a b b c c Оценка репаративной способности клеток методом “ДНК – комет” Площади “комет” были определены как в клетках без повреждающего воздействия, так и в различное время после рентгеновского облучения в дозе Гр. Суммарная площадь “комет” соответствует степени повреждения ДНК исследуемых клеток. Анализ результатов выявил существенные различия между клетками всех исследованных штаммов. Средние суммарные площади “комет” по результатам трех независимых опытов приведены в табл.2. Было показано, что интактные клетки штаммов, полученных от больных АТ (клетки AT6SP и AT8SP) содержат достоверно большее количество повреждений ДНК. Особенно существенно это выражено в клетках AT8SP. Через 30 мин после рентгеновского облучения суммарная площадь “комет” резко возрастала во всех изученных штаммах, что соответствовало появлению в ДНК клеток большого количества разрывов. При этом количество разрывов в клетках больных АТ незначительно (в среднем на 10%), но достоверно превышало таковое как в клетках здорового донора, так и в штаммах, полученных от гетерозиготных носителей заболевания. Способность клеток к воссоединению разрывов ДНК после облучения определялась суммарными площадями “комет” через 2.5 часа после облучения. За этот период времени в клетках больных АТ происходила репарация лишь трети повреждений ДНК, в то время как в клетках здорового донора этот процесс практически завершался. В то же время в клетках гетерозиготных носителей заболевания воссоединение разрывов ДНК проходило быстрее, чем у больных АТ, но не достигал уровня репарации ДНК в клетках здорового донора. Следует отметить, что фибробласты, полученные от больной с клинической формой атаксии, возникшей в результате опухоли мозжечка (клетки AT7SP), демонстрируют также сниженный репаративный потенциал по сравнению с клетками здорового донора и по этому показателю сходны с клетками гетерозиготных носителей АТ.
Название штамма Площадь комет ( S ) клеток без облучения, 30 мин после 2.5 ч после облучения, облучения, VH10 3.00±0.86 83.00±3.90 13.83±5. AT6SP 9.50±5.00 94.50±1.80 72.16±2. AT(F)6SP 9.50±2.18 81.16±6.71 22.66±2. AT(M)6SP 8.66±4.31 78.16±6.80 20.00±0. AT8SP 18.83±2.93 93.33±0.17 74.16±1. AT(M)8SP 6.66±0.76 83.50±1.00 38.16±1. AT(F)8SP 4.50±0.86 85.83±2.84 28.83±2. AT7SP 9.66±1.23 85.50±2.92 32.66±3. Табл. 2. Средние значения площадей комет (S) до и после рентгеновского облучения в дозе 2 Гр.
Статистически достоверные отличия от контроля при p0.05.
На тех же препаратах были подсчитаны апоптозные клетки, которые отличаются от обычных “комет” тем, что у них практически отсутствует «ядро кометы», а вся ДНК сосредоточена в “хвосте”. Эти клетки мы не учитывали при подсчете суммарных площадей “комет”, так как считали их нежизнеспособными. На рис. 8 представлена динамика доли таких клеток в различных исследованных штаммах в интактном состоянии и после рентгеновского облучения. В клетках штаммов больных АТ уровень спонтанного апоптоза существенно выше, чем в клетках здорового донора, и несколько выше, чем в клетках гетерозиготных носителей заболевания.
Количество апоптозных клеток после облучения также существеннее возрастает в клетках больных АТ, чем в клетках здорового донора, клетках гетерозиготных носителей заболевания мы наблюдаем промежуточный результат.
35 Доля клеток, % 3 инт 30 мин 2.5 ч Рис. 8. Анализ изменения доли клеток в апоптозе до и после рентгеновского облучения в дозе 2 Гр.
1 – AT8SP, 2 – VH10, 3 - AT8(F)SP, 4 – AT8(M)SP.
Анализ клеточного цикла методом проточной цитометрии.
В табл.3 и на рис. 9 мы приводим результаты, полученные при исследовании параметров клеточного цикла как при обычном росте клеточных культур, так и после рентгеновского облучения в дозе 5 Гр для штаммов фибробластов больной AT8SP и членов ее семьи, а также больной AT7SP, страдающей атаксией, возникшей в результате опухоли мозжечка. Показано, что клетки здорового донора VH10 имеют ненарушенную систему блокирования клеточного цикла в ответ на повреждение ДНК, что приводит к накоплению этих клеток в фазе G1 и G2 в течение 3 сут после рентгеновского облучения. Клетки больной АТ (AT8SP), напротив, демонстрируют полную радиорезистентность и не изменяют параметров клеточного цикла в течение всего эксперимента. Клетки гетерозиготных носителей мутантного гена atm демонстрируют менее выраженную и длительную остановку клеточного цикла, нежели клетки здорового донора. Сходная картина наблюдается и в клетках больной AT7SP. При сравнении динамики роста клеток исследованных штаммов видно, что клетки здорового донора гораздо сильнее зависят от ростовых факторов среды и быстрее снижают уровень синтеза ДНК, чем клетки больных АТ и гетерозиготных носителей заболевания.
Название Фаза Чис Исход Без 24 ч 2 сут 48 ч 3 сут 72 ч 4 сут штамма клеток клеточн ло ная облуче после роста, после роста, после роста,% ого кле культу ния, 1 облуче % облучен % облуче цикла ток ра, % сут ния, % ия, % ния, % роста% VH10 G0 95.66 47.11 73.56 82.83 81.24 90.72 82.31 90. S 100 0.00 17.69 9.42 9.86 2.44 3.24 2.43 2. G2-M 4.34 35.20 17.02 7.31 16.32 6.04 15.26 6. AT8SP G0 54.88 39.15 44.38 55.72 48.33 52.48 44.96 52. S 100 14.36 26.01 27.74 12.82 21.97 15.02 24.03 30. G2-M 30.76 34.84 27.88 31.46 29.70 32.50 31.01 17. AT(M)8SP G0 80.70 74.20 71.70 75.20 68.76 77.70 66.73 82. S 100 4.70 7.40 4.80 15.40 4.60 8.80 7.58 4. G2-M 14.60 18.40 23.50 9.40 26.64 13.50 25.69 12. AT(F)8SP G0 80.50 65.80 69.57 80.10 64.48 74.20 68.82 64. S 100 5.10 16.70 6.50 12.65 11.74 14.50 10.75 8. G2-M 14.40 17.50 23.93 7.25 23.78 11.30 20.43 27. AT7SP G0 85.70 75.70 70.40 70.40 68.35 64.50 68.90 71. S 100 4.93 20.04 2.94 14.70 11.70 17.00 15.30 11. G2-M 9.37 4.26 26.66 14.90 19.95 18.50 15.80 17. Табл. 3. Распределение клеток по фазам клеточного цикла в течение 4 сут до и после рентгеновского облучения в дозе 5 Гр.
Исх.культура 2 сут роста 3 сут роста 2 сут/обл 3 сут/обл 14.36 12.82 15.02 21.97 24. a 5.10 12.65 14.50 11.74 10. b 0 9.86 3.24 2.44 2. с 0 2c 4c 2c 4c 2c 4c 2c 4c 2c 4c Рис. 9. Зависимость распределения клеток штаммов AT8SP (а), AT(F)8SP (b), VH10 (с) по фазам клеточного цикла до и после рентгеновского облучения в дозе 5 Гр.
По горизонтали – содержание ДНК, ед. плоидности (с);
по вертикали- число клеток. На каждой гистограмме указана доля клеток (%), находящихся в фазе S.
ОБСУЖДЕНИЕ Уменьшение количества гамма-изоформы белка гетерохроматина НР является одним из надежных маркеров старения, пригодным для выявления возрастных изменений как в фибробластах, полученных от пожилых доноров, так и в клетках больных прогерией (Scaffidi, Misteli, 2006;
Смирнова и др., 2008а, 2008б). По этому маркеру атаксия-телеангиэктазия является, безусловно, болезнью преждевременного старения, а гетерозиготное носительство гена данного заболевания ведет к появлению признаков преждевременного старения на клеточном уровне.
Фосфорилированная форма гистона Н2АХ (-Н2АХ) традиционно используется в качестве маркера двунитевых разрывов ДНК. Также было отмечено накопление фосфорилированной формы гистона -Н2АХ в ядрах стареющих клеток в культуре, клетках пожилых доноров или больных прогериями (Sedelnikova et al., 2004;
Scaffidi, Misteli, 2006). Это явление может быть связано как с накоплением нерепарированных DSBs или модификаций хроматина, воспринимаемых как DSBs стресс-киназами АТМ и ДНК-зависимой протеинкиназой (DNA-PK) при повреждении ДНК (Stiff et al., 2004), или протеинкиназой ATR при остановке репликации (Takahashi, Ohnishi, 2005), так и с появлением незащищенных коротких теломер (Hao et al., 2004). Таким образом, накопление фосфорилированной формы гистона -Н2АХ в культуре клеток, в ядрах которых они появляются без действия повреждающих агентов, может быть одним из объективных критериев старения на клеточном уровне (Ayoub et al., 2008: Scaffidi, Misteli, 2006).
Сопоставляя все полученные нами данные с данными литературы, можно сделать вывод, что в соответствии с динамикой выявления фосфорилированной формы гистона -Н2АХ как в интактных клетках, так и после повреждения ДНК все штаммы больных с АТ демонстрируют признаки ускоренного старения и снижения активности репарационных процессов. В клетках гетерозиготных носителей мутантного гена atm выявляется промежуточный между нормой и патологией фенотип.
Белок 53ВР1 является традиционным маркером репарационных процессов (Wilson, Stern, 2008). Его динамика в ответ на повреждение ДНК обычно совпадает с динамикой -Н2АХ, так как они часто колокализуются в зоне конформационных изменений хроматина (двунитевых разрывов ДНК).
Белок 53BP1 после ионизирующего облучения фосфорилируется по серину 1219 (Lee et al., 2008). Этот белок играет также важную роль в остановке клеточного цикла в контрольной точке в ответ на повреждающие воздействия (Iwabuchi et al., 2008;
Eliezer et al., 2009). При сравнении рис. 3, а и б видно, что в разных семьях мы, вероятно, имеем дело с различными мутациями в гене atm.
В фибробластах больной АТ6SP через 24 ч после облучения сохраняется достоверно большее количество фокусов 53ВР1, хотя через 2.5 ч его количество почти не отличалось от такового в контроле (рис.3, а, кривая 4). В штамме АТ8SP основные различия, напротив, отмечаются через 2.5 ч после облучения, а к 24 ч различия практически нивелируются (рис.3, б, кривая 4). Эти данные говорят о нарушении процессов репарации двунитевых разрывов ДНК, но в случае штамма АТ6SP скорее всего эти нарушения затрагивают репарацию с участием гомологической рекомбинации, в случае штамма АТ8SP – негомологичное воссоединение концов ДНК.
Анализ параметров клеточного цикла методом проточной цитометрии позволяет выявить способность клеток к формированию блоков G1/S и G2/M после облучения (Iwabuchi et al., 2008). Данные литературы показывают, что клетки больных АТ демонстрируют радиорезистентность клеточного цикла (Houldsworth, Lavin, 1980;
Beamish, Lavin, 1994;
Beamish et al., 1996;
Hannan et al., 2002) и описан сигнальный путь, необходимый для ее проявления (Bai, Murnane, 2003). Полученные нами результаты об отсутствии блоков клеточного цикла после рентгеновского облучения подтверждают диагноз атаксии телангиэктазии у больных AT6SP и AT8SP и показывают влияние гетерозиготного носительства заболевания на способность формировать блоки клеточного цикла в ответ на облучение.
Исследование белка p21Waf1/Cip1 в клеточных линиях АТ и гетерозиготных носителей мутации в гене atm является еще одним важным этапом в понимании механизма радиочувствительности клеток и решении вопроса о том, является ли данный белок маркером преждевременного старения. Идентифицировано более сотни генов, являющихся мишенями транскрипционной активности р53. Среди них гены, продукты которых регулируют клеточный цикл. Важнейшим из них является белок p21Waf1/Cip1 - ингибитор циклин-зависимых киназ из семейства Cip/Kip (Shi et al., 2008). Повышение его экспрессии вызывает остановку клеточного цикла в поздней G1 фазе, что обусловливается связыванием им комплексов циклин Е/Сdk2, подавлением их способности фосфорилировать белки семейства рRb и высвобождать транскрипционные факторы E2F (Копнин, 2000;
Radhakrishnan et al., 2006, Hill et al., 2008). В клетках больных АТ многие процессы, наблюдаемые в клетках здорового донора после рентгеновского облучения, либо происходят с существенной временной задержкой, например, стабилизация белка р53, либо вовсе полностью подавлены, например, остановка клеточного цикла (Спивак и др., 2005). Таким образом, уменьшенное количество Р21 в клетках больных АТ соответствует снижению количества фосфорилированного Р53 в этих клетках, с этим же связана и радиорезистентность клеточного цикла при АТ. Тот факт, что у гетерозиготных носителей данного заболевания достоверные отличия не выявляются в отличие от клеток здорового донора, это не позволяет выявлять гетерозиготное носительство мутантного гена atm с помощью белка Р21, но дает возможность анализировать процессы повышенной радиочувствительности и преждевременного старения на клеточном уровне. Вероятно, одного здорового аллеля гена достаточно для появления детектируемого количества белка Р21, но недостаточно для нормальной регуляции клеточного цикла. Вероятно, такое нарушение регуляции клеточного цикла связано в первую очередь не с Р21, а с какими-то другими АТМ-зависимыми факторами, например, с Е2F.
Анализ количества повреждений ДНК, в первую очередь – двунитевых разрывов (DSBs), после действия генотоксических агентов является одним из наиболее распространенных подходов для изучения способности клеток к репарации. Методом “ДНК-комет” было показано, что количество образующихся разрывов ДНК зависит от дозы и природы повреждающего агента, а способность различных типов клеток воссоединять разрывы - от активности их систем репарации и от времени (Kolman et al., 1997;
Chovanec et al., 1998).
Под действием ионизирующей радиации двунитевые разрывы ДНК (DSBs) в диплоидных фибробластах, полученных от здорового донора, формируются в первые минуты после повреждения и репарируются на 90 % менее чем за 4 ч (Mirzayans et al., 2006), что соответствует данным, полученным нами для штамма нормальных фибробластов человека VH10. Клетки больных АТ демонстрируют существенную задержку репарации, через 2.5 ч после облучения соединяются лишь 20-30 % разрывов. Это подтверждают данные о снижении активности процессов репарации в клетках больных АТ, полученные ранее в нашей лаборатории с использованием других методов (Хамасуридзе и др., 1999, Спивак и др., 2005, Смирнова и др., 2008). Динамика воссоединения разрывов после рентгеновского облучения, описанная нами с использованием метода “ДНК – комет”, соответствует той, что мы наблюдали, применяя метод непрямой иммунофлуоресценции для определения количества фокусов белка Н2АХ и белка 53ВР1 (Полуботко и др., 2009). Восстановление поврежденной ДНК, о котором мы можем судить по элиминации фокусов белка -Н2АХ и белка 53ВР1 в ядрах клеток, у больных АТ за 24 ч происходит лишь наполовину, в то время как в клетках здорового донора репарация ДНК к этому времени практически завершается. В клетках гетерозиготных носителей мутантного гена atm процессы репарации ДНК были достоверно замедлены, но не так резко, как у больных АТ.
В литературе было описано, что количество спонтанных повреждений ДНК в клетках увеличивается с возрастом (Sirota, Kuznetsova, 2008). Также известно, что при обработке повреждающими агентами фибробласты более старых животных чаще уходят в апоптоз (Анисимов и др., 2003). Наши данные показывают, что уровень спонтанных нарушений ДНК и апоптоза в интактных клетках АТ превышает уровень таковых в контроле в 3 раза (клетки АТ6SP) и в 6 раз (клетки AT8SP). В то же время интактные клетки гетерозиготных носителей мутантного гена также демонстрируют повышенный, но менее выраженный, чем у больных АТ, уровень спонтанных повреждений ДНК. Ранее при исследовании клеток, полученных от больных АТ и их кровных родственников – гетерозиготных носителей заболевания - нами было показано, что клеточные маркеры старения выявляются с более высокой частотой как в клетках самих больных АТ, так и в клетках гетерозиготных носителей заболевания (Полуботко и др., 2009). Сопоставляя полученные нами данные с данными литературы, можно отметить, что штаммы больных АТ демонстрируют признаки ускоренного старения и снижения активности репарационных процессов на клеточном уровне, а в клетках гетерозиготных носителей данного заболевания выявляется промежуточный фенотип. Все это в совокупности подтверждает наше представление об атаксии-телеангиэктазии как о синдроме преждевременного старения, а не только повышенной радиочувствительности.
Крайне интересным представляется анализ клеток больной AT7SP, у которой атаксия является результатом опухоли мозжечка. В то же время у этой больной по сравнению с клетками здорового донора понижена способность к репарации разрывов ДНК и выявляется повышенное количество маркеров старения, так же как у гетерозиготных носителей АТ. Способность к формированию блоков клеточного цикла в ответ на рентгеновское облучение у больной AT7SP также снижена. При анализе этого клинического случая нужно учитывать, что уровень радиочувствительности клеток больных АТ столь широк, что позволил выделить особую группу больных, диагностированных как АТ-вариант (Gilad et al., 1998). У таких пациентов наблюдались явные признаки заболевания, но чувствительность клеток к ионизирующей радиации по ряду признаков была выражена слабее, чем при классической форме АТ (Taylor et al., 1987). Ранее в нашей лаборатории были описаны клетки, отнесенные нами к АТ-варианту (клетки AT1SP, AT2SP) (Спивак и др., 2005). К сожалению, подробный анализ этих клеток методом проточной цитометрии и “ДНК-комет” не проводился, как и анализ этих штаммов на наличие маркеров старения. Совместный анализ всех этих данных позволяет предположить, что штамм AT7SP может являться как гетерозиготным носителем АТ или больной АТ-вариант, так и гетерозиготным носителем дефекта какого-либо другого гена, вовлеченного в репарацию двунитевых разрывов ДНК (напрмер, ATR или ligIV). В любом случае, у этой больной есть повышенная предрасположенность к развитию опухолей и преждевременному старению, что необходимо учитывать при терапии.
ВЫВОДЫ 1. Атаксия-телеангиэктазия является прогероидным синдромом, а гетерозиготы имеют черты преждевременного старения на клеточном уровне.
2. Способность к репарации двунитевых разрывов, определяемая методом “ДНК-комет” и выявлением фокусов -Н2АХ и 53ВР1, достоверно снижена в клетках больных АТ и гетерозиготных носителей заболевания.
3. Клетки больных АТ не способны формировать остановку клеточного цикла после облучения. У гетерозиготных носителей формирование блоков клеточного цикла после облучения нарушено.
4. Р21 не может служить маркером для определения гетерозиготного носительства АТ.
5. Клетки больной AT7SP обладают повышенной радиочувствительностью неясной этиологии, что может являться причиной развития у нее опухоли в раннем возрасте.
Публикации по теме диссертации Полуботко Е.А., Смирнова Н.В., Плескач Н.М., Михельсон В.М., Спивак И.М.
2009. Особенности преждевременного старения при атаксии-телеангиэктазии.
Цитология. 51 (8): 712-718.
Полуботко Е.А., Шатрова А.Н., Плескач Н.М., Михельсон В.М., Спивак И.М.
2009. Клеточный репаративный потенциал в семьях больных атаксией телеангиэктазией. Цитология. (12): 1036 – 1041.
2008. Определение наследственной радиочувствительности Полуботко Е.А.
при атаксии-телеангиэктазии и немедуллярном раке щитовидной железы.
Тезисы конференции “Биология: от молекулы до биосферы”. Харьков:125.
Полуботко Е.А., Шатрова А.Н., Зимарина Т. В., Спивак И.М., Михельсон В.Н.
2008. Определение наследственной радиочувствительности в семьях больных с синдромом атаксия-телеангиэктазия и в семьях больных немедуллярным раком щитовидной железы. Тезисы конференции молодых ученых “Биология – наука ХХ века”. Москва: 115.
Михельсон В.М., Полуботко Е.А., Шатрова А.Н., Смирнова Н.В., Плескач Н.М., Спивак И.М. 2007. Наследственная радиочувствительность и преждевременное старение в семьях больных атаксией-телеангиэктазией и немедуллярным раком щитовидной железы. Тезисы конференции “Фундаментальные науки медицине”. Москва: 186.
Михельсон В.М., Спивак И.М., Зимарина Т.В., Полуботко Е.А., Томилин Н.В.
2007. Выявление повышенной наследственной радиочувствительности с помощью оценки репаративного потенциала на клеточном уровне. Тезисы докладов конференции “Фундаментальные науки-медицине”. Москва: 158.
Список цитируемой литературы Анисимов В.Н. 2003. СПб.: Наука. Кольман А. 1997.. Цитология. 39 (6): 420 434. Копнин Б.П. 2000. Биохимия. 65 (1): 5-33. Михельсон В. М. 1996.
Клиническая геронтология. (4): 29-35. Наседкина Т.В., Кузминова А.Е., Сурков С.А., Плескач Н.М., Прокофьева В.В., Спивак И.М., Михельсон В.М., Полетаев А.И. 1997. Цитология. 39: 809-821. Полуботко Е.А., Смирнова Н.В., Плескач Н.М., Михельсон В.М., Спивак И.М. 2009. Цитология. 51 (8): 712-718. Смирнова Н.В., Спивак И.М., Плескач Н.М., Жеребцов С.В., Аксенов Н.Л., Михельсон В.М.
2008а. Цитология. 50 (10): 868 – 877. Смирнова Н.В., Спивак И.М., Плескач Н.М., Михельсон В.М. 2008б. Цитология. 50 (9): 780-788. Спивак И. М., Плескач Н. М., Боотсма Д., Кольман А. 1997. Цитология 39(6): 420-434.Спивак И.М.
1999. Цитология. 41 (5): 338-380. Спивак И.М., Смирнова Н.В., Плескач Н.М., Ледащева Т.А., Михельсон В.М. 2005. Цитология. 47 (10): 898-906. Спивак И.М., Плескач Н.М.,Смирнова Н.В., Шатрова А.Н., Михельсон В.М. 2007.
Технологии живых систем. 4(5-6):39-54.Хомасуридзе М. М., Спивак И. М., Плескач Н. М., Михельсон В. М. 1999. Цитология.41 (5): 412-419.
Abraham R. T. 2001. Genes Dev. 15: 2177–2196.Ayoub N., Jeyasekharan A.D., Bernal J.A., Venkitaraman A.R. 2008. Nature. 453: 682-686.Bai Y., Murnane J.P.
2003. Mol Cancer Res. 1(14):1058-69.Barenfeld L. S., Nergadze S. G., Pleskach N.
M., Prokofjeva V. V., Mikhelson V. M. 1998. Mutat. Res. 408(3):219-226. Beamish H., Lavin M.F. 1994. Int J Radiat Biol 65:175–184.Beamish H., Williams R., Chen P., Lavin M. 1996. J Biol Chem 271:20486–20493.Belyayev I. Y., Spivak I. M., Kolman A., Harms-Ringdahl M. 1996. Mutat. Res. 358 : 223-230. Chovanec M., Naslund M., Spivak I., Dusinska M., Cedervall B. and A. Kolman. 1998. Environment.
Mol. Mut. 32 (3):223-228. Christmann M., Tomicic M.T., Roos W.P., Kaina B. 2003.
Toxicology.193: 3-34.Eliezer Y., Argaman L., Rhie A., Doherty A.J., Goldberg M.
2009. J. Biol. Chem. 284: 426-435.Fei P. and El-Deiry W.S. 2003. Oncogene.
22(37): 5774-5783.Gilad S., Chessa L., Khosravi R., Russell P., Galanty Y., Piane M., Gatti R.A., Jorgensen T.J., Shiloh Y., Bar-Shira A. 1998. Am J Hum Genet.
62(3):551-61.Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Lbrich M., Jeggo P.A. 2008. Mol. Cell. 31: 167-177.Hanawalt P.S. 2001. Nature. 485: 3 13.Hannan MA, Hellani A, Al-Khodairy FM, Kunhi M, Siddiqui Y, Al-Yussef N, Pangue-Cruz N, Siewertsen M, Al-Ahdal MN, Aboussekhra A. 2002. Carcinogenesis.
23(10):1617-24. Hao L.Y., Strong M.A., Greider C.W. 2004. J. Biol. Chem. 279:
45148-45154.Hill R., Bodzak E., Blough M.D., Lee P.W. 2008. Cell Cycle. 7: 2535 2543.Hoeijmakers J.H. 2001. Nature. 411: 366-374.Houldsworth J., Lavin M.F.
1980. Nucleic Acids Res 8:3709–3720.Huang L., Shyder A.R., Morgan W.F. Oncogene. 22 (37): 5848-5854.Iliakis, G., Wang, Y., Guan, J., Wang, H. 2003.
Oncogene 22:5834-5847.Iwabuchi K., Matsui T., Hashimoto M., Matsumoto Y., Kurihara T., Date T. 2008. Biochem. Biophys. Res. Commun. 376: 509-513.Jeggo P. A., Carr A. M., Lehmann A. R. 1998. Trends Genet. 14: 312–316.Karlsson K.H., Stenerlow B. 2004. Radiat. Res. 161: 517-527.Kim S.T., Xu B., Kastan M.B. 2002.
Genes Dev. 16(5):560-70. Kolman A., Spivak I., Naslund M., Dusinska M., Cedervall B. 1997. Enviroment. Mol. Mutagen. 30: 40-46.Kuo L.J., Yang L.X. 2008. In Vivo.
22: 305-9. Lansdorp PM. 2000. Mech Ageing Dev. 118(1-2):23-34. Lavin M.F. 2008.
Nat. Rev. Mol. Cell Biol. 9: 759-69.Lee H., Kwak H., Cho I.T., Park S.H., Lee C.H.
2008. Biochem Biophys Res Commun. 378: 32-6. Lin W.C., Lin F.T., Nevins J.R.
2001. Genes Dev. 15(14):1833-44. Mikhelson V. M. 2001. Mechanisms of Ageing and Development. 122 (13): 1361-1365.Mirzayans R., Severin D., Murray D. 2006.
Int J Radiat Oncol Biol Phys. 66(5):1498-505.Nakayama H. 2002. Oncogene.
21(58):9008-21. Nygren J., Cedervall B., Ericksson M., Dusinska M., Kolman A.
1994. Envir. Mol. Mutagen. 24: 161-167.Platzer M., Rotman G., Bauer D., Uziel T., Savitsky K., Bar-Shira A., Gilad S., Shiloh Y., Rosenthal A. 1997. Genome Res.
7(6):592-605.Pulverer B. 2003. Nature Cell Biol. 5:96.Radhakrishnan S.K., Gierut J., Gartel A.L. 2006. Oncogene. 25: 1812-1815.Sancar A., Lindsey-boltz L.A., Unsal Kamaz K., Linn S. 2004. Annu.Rev.Biochem. 73: 39-85.Sandoval N., Platzer M., Rosenthal A., Doerk T., Bendix R., Skawran B., Stuhrmann M., Wegner R.-D., Sperling K., Banin S., Shiloh Y., Baumer A., Bernthaler U., Sennefelder H., Brohm M., Weber B., Schindler D. 1999. Hum. Mol. Genetics. 8 (1): 69-79.Sano M., Minamino T., Toko H., Miyauchi H., Orimo M., Qin Y., Akazawa H., Tateno K., Kayama Y., Harada M., Shimizu I., Asahara T., Hamada H., Tomita S., Molkentin J.D., Zou Y., Komuro I. 2007. Nature. 446(7134):444-8.Scaffidi P., Misteli T. 2006.
Science. 312: 1059 – 1063.Sedelnikova O.A., Horikawa I., Zimonjic D.B., Popescu N.C., Boner W.M., Barrett J.C. 2004. Nat. Cell Biol. 6: 168-170.Shi Q., Wang X., Ren J. 2008. Biophys. Chem. 138: 138-43.Shiloh Y. 2003. Nature Rev. 3: 155 168.Sirota N.P., Kuznetsova E.A. 2008. Bull Exp Biol Med. 145(2):194-7.Stiff T., O’Driscoll M., Rief N., Iwabuchi K., Lobrich M., Jeggo P.A. 2004. Cancer Res. 64:
2390-2396.Takahashi A., Ohnishi T. 2005. Cancer Lett. 229: 171-179.Tanaka T., Halicka H.D., Huang X., Traganos F., Darzynkiewich.Z. 2006. Cell cycle. 5: 1940 1945.Taylor A.M., Flude E., Laher B., Stacey M., McKay E., Watt J., Green S.H., Harding A.E. 1987. J Med Genet. 24(11):669-77.Thaker J., Zdzienicka M.Z. 2005.
DNA Repair. 4 (2): 303-314.Uziel T., Savitsky K., Platzer M., Ziv Y., Helbitz T., Nehls M., Boehm T., Rosenthal A., Shiloh Y., Rotman G. 1996. Genomics. 2: 317-320.Ward I.M., Chen J. 2001. J.Biol.Chem. 276 : 42462-42467.Westphal C., Rowan S., Schmaltz C., Elson A., Fisher D., Leder P. 1997. Nat. Genet. 16 : 397-401.Wilson K.A., Stern D.F. 2008. Cell Cycle. 7: 3584-3594. Zhou B.-B.S., Bartek, J. 2004.
Nature Reviews. 4 : 216-225.