
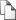
Определение регуляторных сегментов в геномах методами теоретического анализа последовательностей нуклеотидов днк
На правах рукописи
Макеев Всеволод Юрьевич Определение регуляторных сегментов в геномах методами теоретического анализа последовательностей нуклеотидов ДНК 03.00.02 Биофизика
АВТОРЕФЕРАТ
диссертации на соискание ученой степени доктора физико-математических наук
Москва – 2009 1
Работа выполнена в лаборатории биоинформатики ФГУП «Государственный научно-исследовательский институт генетики и селекции промышленных микроорганизмов ГосНИИгенетика».
Научный консультант: Доктор физико-математических наук, профессор Туманян Владимир Гаевич
Официальные оппоненты:
Доктор физико-математических наук, профессор, член-корреспондент РАН Гурский Георгий Валерьянович Доктор физико-математических наук Намиот Владимир Абрамович Доктор биологических наук, профессор Озолинь Ольга Николаевна
Ведущая организация: Институт теоретической и экспериментальной биофизики РАН
Защита состоится «22» октября 2009 г. в 15 ч. 30 мин на заседании диссертационного совета Д 501.002.11 при Московском Государственном Университете им. М.В.Ломоносова по адресу 119991, ГСП-1, Москва, Ленинские горы, МГУ им. М.В.Ломоносова, физический факультет, аудитория 5-19.
С диссертацией можно ознакомиться в библиотеке физического факультета МГУ
Автореферат разослан 21 сентября 2009г.
Ученый секретарь диссертационного совета Д 501.002. доктор физико-математических наук Г.Б. Хомутов
Общая характеристика работы
Актуальность проблемы В результате быстрого прогресса экспериментальной техники современная биология подошла вплотную к решению одной из своих фундаментальных проблем, а именно – проблемы понимания того, как происходит реализация наследственной информации в живом организме. Решение этой важнейшей проблемы современных генетики и биофизики должно повлечь за собой успехи в ряде практических задач в областях биотехнологии и медицины. К ним, в частности, относится управление дифференцировкой тканей, выращенных в культуре;
понимание роли конкретных аллелей в возникновении заболеваний, имеющих наследственный компонент, а также производство лекарств белковой природы в культурах клеток млекопитающих, модифицированных методами генной инженерии.
Деление и дифферецировка клеток контролируются тысячами актов взаимодействий между макромолекулами белков и нуклеиновых кислот.
Современные экспериментальные технологии позволяют получить огромные объемы экспериментальных данных, характеризующих взаимодействие биологических макромолекул в различных условиях. Одной из непосредственных задач современной биофизики является использование вычислительных физических методов для систематизации и сопоставления данных, полученных различными экспериментальными методами. За всю свою историю научные исследования никогда не располагали средствами такой мощи для переработки информации и никогда не сталкивались с необходимостью переработки информации такого объема, приходящией из различных источников и открытых для общего доступа.
Компьютерная биофизика, по существу, становится полигоном для применения сложных статистических методов анализа данных и оценки гипотез. Основой работы является использование вычислительных методов для анализа тех закономерностей в строении последовательностей нуклеотидов в ДНК, которые связанны со структурно-физическими основами регуляции инициации транскрипции. Объекты исследования – участки геномов эукариот. Выбор такого объекта имеет ряд технических преимуществ. Во-первых, тексты ДНК дискретны и при современном уровне технологии секвенирования число ошибок в последовательностях не превышает, как минимум, одной ошибки на пять тысяч оснований (Robertson.G., et al. (2007)). Поскольку специфичность ДНК-белкового распознавания значительно ниже, при решении задач, описанных в настоящей работе, этим уровнем ошибок можно пренебречь. Во-вторых, секвенирование сейчас относительно дешево, а поэтому в открытом доступе имеются последовательности геномов тысяч видов прокариот и сотен видов эукариот.
В третьих, геном является одномерной струтурой, поэтому молекула гетерополимера ДНК может анализироваться как текст, составленный из символов – мономеров разных типов нуклеотидов. Анализ на уровне текста ДНК позволяет понять большое количество биофизических по существу эффектов, связанных с инициацией транскрипции на молекулярном уровне.
Важной задачей настоящей работы является нахождение участков ДНК, участвующих в работе механизмов, управляющих переключением генов, так как для решения вопросов, связанных с пространственной структурой и физикой взаимодействия элементов регуляторных комплексов прежде всего надо знать какие именно сегменты ДНК несут регуляторную нагрузку и какие факторы белковой природы действуют на эти регулятороные сегменты, вызывая переключение экспрессии конкретных генов.
В работе используется широкий арсенал математических методов анализа последовательностей. В частности, это методы сравнительного анализа последовательностей, грамматический анализ, т.е. анализ структруктурных закономерностей в последовательностях, распознавание характерных образов в последовательностях, а также оценка надежности найденных закономерностей путем построения статистических критериев.
Цель и задачи исследования Целью работы является установление физических основ реализации генетической информации на этапах образования регуляторных комплексов ДНК-белок и функционирования компонентов генома. Это включает в себя:
установление физических характеристик участков последовательности ДНК, несущих регуляторную функцию, и получение распределения участков специфического связывания регуляторных белков в пределах этих регуляторных участках ДНК;
также предполагается установление связи характеристик распределения участков связывания регуляторных белков с физическими свойствами формирующихся иерархически организованных структур ДНК-белковых комплексов.
В работе решались следующие частные задачи:
1. Установление участков ДНК, вступающих в специфическое взаимодействие с белковыми факторами, регулирующими транскрипцию, с помощью специально разработанных методов.
2. Построение формального критерия наличия заданных структур в последовательности нуклеотидов ДНК, на основые вычисления статистической значимости.
3. Построение метода крупномасштабной сегментации последовательности на участки, однородные по своему нуклеотидному составу, с помощью вычисления статистической суммы всевозможных разбиений последовательности на сегменты.
4. Построение метода, позволяющего определять участки ДНК, вступающие в специфическое взаимодействие с белками, путем установления последовательностей нуклеотидов со значительной позиционно специфической энергией взаимодействия с учетом симметрии структуры ДНК.
5. Установление связи между характером распределения ДНК связывающих областей в регуляторных зонах, типами пространственных структур, диктуемых такими распределениями, и функционированием определенных сегментов генома.
6. Разработка способа, позволяющего выделять регуляторные сегменты ДНК (промоторов и энхансеров) как участки ДНК, имеющие высокую аффинность взаимодействия с белками, в том числе и кооперативно связающимися белковыми факторами.
7. Определение регуляторных сегментов ДНК для системы генов, участвующих в раннем развитии Drosophila melanogaster.
Научная новизна и практическая ценность работы В работе впервые получены следующие результаты:
На основе разработаных методов, позволяющих выделять в нуклеотидной последовательности геномной ДНК участки, специфически взаимодействующие с белками-регуляторами транскрипции, установлено характерное взаимное расположение таких участков ДНК, позволяющее обеспечить самоорганизацию нативного инициаторного комплекса при превышении пороговой концентрации регуляторных факторов.
Для тех случаев, когда задача вычисления вероятности появления наблюдаемой конфигурации мотивов в случайной последовательности ДНК не имеет аналитического решения (наблюдаются группы перекрывающихся мотивов, распознаваемых разными факторами) построен алгоритмический метод, позволяющий быстро и точно вычислять статистическую значимость появления наблюдаемой конфигурации мотивов.
Разработан метод сегментации генома на участки со стабильным нуклеотидным составом, в пределах которых допустимо использование одной и той же статистической модели. Метод основан на вычислении статистической суммы по всевозможным разбиениям последовательности на формально-однородные сегменты.
Для определения конкретных участков ДНК, специфически распознаваемых регуляторными белками, создан метод построения множественного локального выравнивания фрагментов ДНК, позволяющий учитывать предполагаемую структуру регуляторного участка.
Создан банк данных мотивов в ДНК, распознаваемых различными регуляторными белками, участвующими в регуляции генов, управляющих ранним развитием Drosophila melanogaster.
Показано, что регуляторные модули, в частности энхансеры, имеют структуру, способствующую кооперативному связыванию регуляторных белков с этими модулями. Этот феномен позволяет идентифицировать регуляторные модули в геноме. Найден ряд новых регуляторных модулей в D. melanogaster.
Основные положения, выносимые на защиту 1. Расположение дистальных регуляторных элементов, в частности энхансеров в геноме может быть определено исходя из наличия в них участков, специфически взаимодействующих с регуляторными белками.
2. Периодические закономерности в расположении участков связывания регуляторных белков в пределах регуляторных модулей могут быть эффективно использованы для предсказания стабильной конформации ДНК белкового инициаторного комплекса.
3. Для регуляторных белков, связывающихся с ДНК в форме димера, удается существенно улучшить распознавание участков ДНК, взаимодействующих с регуляторным белком, путем учета симметрии их структуры.
4. Статистическая значимость наблюдения мотива связывания белков или конфигурации мотивов может быть вычислена точно, путем комбинаторного анализа с помощью модификации алгоритма Ахо-Корасик.
5. Распределение мотивов в последовательности ДНК позволяет судить об уровнях организации генома, которые ответственны за конкретные структурные и регуляторные функции.
Практическое значение работы Практическая значимость работы выражается в том, что разработанные в данном исследовании методы применяются в области биотехнологии: это – разработка эффективных конструктов из культур клеток тканей эукариот, включая млекопитающих;
и в биомедицине: это – связь аллельных вариантов, локализованных в регуляторных областях, с возможными патологиями.
Кроме того, в качестве практических достижений можно назвать создание конкретных программных инструментов, используемых научным сообществом. Такими инструментами являются:
Программа BASIO, позволяющая осуществлять сегментацию хромосомы на участки с определенным характерным составом.
Программа SeSiMCMC построения множественного локального выравнивания последовательностей при условии заданной симметрии выровненной последовательности и определения характерного мотива в выровненном участке [http://bioinform.genetika.ru/SeSiMCMC/].
Программа поиска сегментов ДНК, содержащих кластеры участков связывания одного и того же регуляторного белка (гомотипические кластеры сайтов связывания).
Программа AhoPro, позволяющая оценивать статистическую значимость кластеров мотивов в ДНК. [http://bioinform.genetika.ru/AhoPro].
База данных DMMPMM, содержащая мотивы, распознаваемые белками, регулирующими транскрипцию в ходе раннего развития Drosophia melanogaster [http://line.imb.ac.ru/DMMPMM/].
О результативности практических приложений свидетельствует использование наших результатов в нижеперечисленных исследованиях.
Программа SeSiMCMC успешно принимала участие в международном конкурсе аналогичных программ, проводимом Вашингтонским Университетом (США) [Tompa et. al. //Nat.Biotech (2005), 123, p. 137.], и применялась в ряде исследований совместно с широко используемым комплексом программ GenomeExplorer [Миронов А.А. и др.// Молекул.
биология, 34, 253]. Приоритет работ, с участием автора, в которых была предложена идея кластеризации сайтов связывания в регуляторных элементах [Papatsenko D.A. et al. // Genome Research 2002, 12:470;
Lifanov A.P. et al // Genome Research, 2003,13:579;
Makeev V.J. et al // Nucleic Acids Res. 31:6016] как основного фактора, обеспечивающего сложную регуляцию тканеспецифической экспрессии генов высших эукариот, признан международным научным сообществом. Следует отметить, что упомянутые практические успехи стали возможны потому, что удалось решить ряд теоретических проблем, в частности переформулировать задачу распознавания функциональных участков как задачу распознавания одномерных текстовых образов.
Апробация работы Материалы диссертации докладывались на международных и всероссийских конференциях и семинарах, в том числе: Московском семинаре по компьютерной генетике (Москва, 1997);
Отчетной конференции по программе «Геном человека», (Черноголовка, 1997);
международном семинаре «Mathematical applications in biological sciences» (Tronheim, Norway, 1997, Rouen, France, 1998);
международной конференции по алгоритмам в вычислительной биологии RECOMB 1998 (Lyon, France);
Международной конференции JOBIM 1997 (Montpellier, France);
II, III Съездах биофизиков России (Москва, 1999, Воронеж, 2004), III, IV,V, Международных конференциях по биоинформатике регуляции структуры генов (Новосибирск, 2002, 2006, 2008), I и II Московских международных конференциях по вычислительной биологии (2003, 2005), Международном семинаре ASSCG (Evry, France, 2003), Энгельгардтовской конференции по молекулярной биологии (Суздаль, 2004);
Gordon Research Conference in Human Genetics and Genomics (Newport, USA, 2005), III и IV Съездах биохимиков и молекулярных биологов (Санкт-Петербург, 2005, Новосибирск 2008);
международном семинаре Statistical semantics of genomes (Evry, France, 2008);
Международной выставке Biotechnica 2008 (Hannover, Germany);
Конференции общества Гельмгольца (Москва, 2008);
Российско-Индийских школах-конференциях по биоинформатике и геномике (Хайдарабад, 2006, Новосибирск, 2008) и др.
С использованием материалов диссертации автором сделаны: доклад в Rockefeller University, New York, USA (1999);
два доклада в Georgia Tech, Atlanta, USA (2003, 2004);
доклад в РАН (Москва, 2004);
доклад в Каролинском университете (Стокгольм, 2005) и ряд других выступлений.
Публикации По материалам диссертации опубликовано 36 статей в реферируемых научных журналах (из них 35 в соавторстве), а также более 50 тезисов докладов (см. пред. раздел).
Структура работы Диссертация состоит из семи глав, выводов и библиографии ( наименования). Ее полный объем составляет 184 страницы, количество рисунков 17, количество таблиц 6.
Глава 1. Введение.
Во Введении обосновывается актуальность работы, ее научная новизна, практическая значимость. Описаны цели и задачи исследования, а также практические результаты. Сформулированы положения, выносимые на защиту. Приведена информация об апробации работы, количество публикаций автора, дана структура дисертации.
Глава 2. Анализ нуклеотидых последовательностей регуляторных сегментов ДНК и задача об управлении экспрессией генов в эмбриональном развитии D. melanоgaster.
Данная глава является обзором литературы. В этой главе также формулируются конкретные задачи, решение которых описывается в последующих главах диссертации.
В результате успехов геномных проектов стало понятно, что виды, сильно отличающеся по морфологии, могут иметь весьма сходных набор структурных генов. Эти морфологические различия по-видимому связаны с различиями в регуляции экспрессии генов. В наибольшей степени на морфологии должны сказываться различия в экспрессии генов, управляющих течением эмбрионального развития. Механизмы, контролирующие экспрессию генов, пока изучены недостаточно. Известно, однако, что для инициации транскрипции неоходимо формирование масштабных надмолекулярных структур, включающих в себя многочисленные молекулы белков, в частности, факторов инициации транскрипции, и различные участки молекул ДНК. Возникает вопрос, какие биофизические механизмы отвечают за формирование и стабилизацию этих структур. Очевидно, что эти механизмы должны действовать на нескольких уровнях иерархии макромолекулярных взаимодействий: участки непосредственного контакта, характерные структурные домены макромолекул (спирали и тяжи), а также формирование собственно надмолекулярных структур.
Поскольку информация о структуре очень мала, то единственная возможность получить информацию о физических взаимодействиях, лежащих в основе инициаторного комплекса, заключается в изучение закономерностей в последовательности нуклеотидов ДНК. Одномерная текстовая закономерность в расположении нуклеотидов должна являться проекцией трехмерной структуры макромолекулярной укладки. Реальная природа физических взаимодействий трехмерна, поэтому объяснение текстовых закономерностей в строении последовательностей возможно только на уровне трехмерных явлений. Поскольку макромолекулярный комплекс образует иерархическую структуру, в проекции на одномерную последовательность нуклеотидов ДНК эта структура проявляется как иерархия закономерностей в последовательности, существующих на разных масштабах. Очень часто единственным экспериментальным источником информации является последовательность ДНК. Поэтому встает вопрос: «До какой степени иерархия физических явлений на разных масштабах длин, лежащая в основе формирующегося ДНК-белкового комплекса, может быть оценена исходя только из последовательности ДНК». Решению этого вопроса посвящена настоящая работа.
Рядом исследователей, включая автора, в конце 90х годов было сделано наблюдение, что сегменты ДНК, контролирующие транскрипцию генов (цис регуляторные модули или ЦРМ), часто содержат большое число сходных подпоследовательностей длиной от единиц до нескольких десятков нуклеотидов. В некоторых случаях было показано, что эти «перепредставленные слова» в нуклеотидном тексте взаимодействуют с белками, регулирующими транскрипцию этих генов (транскрипционными факторами, ТФ). Таким образом определяя местоположение сайтов связывания ТФ (ССТФ), а также анализируя распределение ТФ в пределах регуляторных модулей, в особенности распределение расстояний между разными ССТФ, можно получить информацию, о том, какие физические свойства ДНК-белкового комплекса важны для его фукнционирования.
Используя полученную информацию сделать выводы о строении трехмерных макромолекулярных комплексов на разных масштабах длин. Полученная информация позволит предсказать ССТФ и ЦРМ, неизвестные из прямых экспериментальных данных.
В обзоре литературы также приведены основные открытые ресурсы, содержащие экспериментальные данные по развитию Drosophila, регуляции экспрессии различных генов Drosophila, а также белок-белковому и ДНК белковому взаимодействию у Drosophila и других видов.
Глава 3. Использование метода регулярных языков для вычисления математического ожидании и дисперсии числа мотивов, встреченных в случайной последовательности данной длины, моделирующей ДНК.
Существенным элементом структуры ДНК, определяющим регуляторные свойства данного района генома, являются участки специфического связывания регуляторных белков. Математически эти участки несут сигналы, формализованные как «мотивы».
Предметом этой главы является теоретическое вычисление математического ожидания и дисперсии количества случайных появлений одного и того же мотива в случайном тексте. Основной целью данного раздела является оценка применимости приближения Пуассона путем вычисления точного значения дисперсии для числа встреченных мотивов в случайной последовательности.
Все последовательности длины n, содержащие N вхождений мотива H представляют собой регулярный язык (Guibas, Odlyzko, 1981;
Regnier, Szpankowski, 1998;
Regnier, 2000). Для вычисления матожидания и дисперсии N этот язык представляется как комбинация сумм и произведений более простых языков, каждый из которых содержит не более одного вхождения мотива H. Наиболее удобным оказывается выбор языков-компонент, при котором все вхождения слов H i возникают при конкатенациях языков компонент, и добавление каждого компонента приносит единственное вхождение H i. Каждый такой язык M ij называется минимальным, он содержит все такие слова, что каждое слово, принадлежащее языку произведению H i M ij содержит единственное вхождение H j в качестве суффикса и не содержит других вхождений H, кроме префикса H i и суффикса H j. Набор минимальных языков M ij, дополненный языками первого появления Ri (оканчивающимися на H i ) и терминальными языками U i (не порождающими вхождений H ни с какими префиксами H i ) достаточен для того, чтобы записать все последовательности произвольной длины, содержащие фиксированное число r вхождений мотива H. В матричном виде это записывается как S r = RM r-1U*, где R - вектор-строка начальных языков, компоненты которого заканчиваются на различных вхождениях мотива H i, M – матрица минимальных языков M ij, а U* – вектор-столбец терминальных языков U i.
Для определения статистических характеристик числа мотивов, встреченных в случайной последовательности длины n, используется аппарат производящих функций. (Guibas, Odlyzko, 1981;
Regnier, Szpankowski, 1998;
Regnier, 2000). Производящая функция – это формально сопоставленный регулярному языку ряд по q + 1 переменным, где q - количество слов в мотиве H:
( z, u,..., u ) = z P ( N ( H ) = r,..., N ( H ) = r ) u r...u q, r n FN H,..., N H 1 q 1 1 q q 1 q n r1,..., rq в котором степень z n стоит при каждом элементе-последовательности длины n, а степень u ri стоит при каждом элементе, содержащем ri вхождений слова H i. Коэффициенты ряда – вероятности элементов-последовательностей, вычисленные в рамках принятой случайной модели. Обозначая символом z n член при степени z n, можно записать связь между значением производящей функции и математическим ожиданием и дисперсией числа встреченных в случайной последовательности вхождений мотива H i. При этом математическое ожидание и дисперсия количества наблюдающихся вхождений мотива H вычисляется следующим образом:
q E ( N ( H )) = E ( N ( Hi )) ;
i = q q ( ) V ( N ( H ) ) = V ( N ( H i ) ) + Cov N ( H i ), N ( H j ) ;
i j i = i, j = F ( z, u ) F ( z, u ) ( ) Cov N ( H i ) N ( H j ) = z n u u F ( z, u ) u.
u j u = i j i Таким образом, для вычисления дисперсии количества мотивов, встреченных в случайной последовательности длины n необходимо вычислить n -й член ряда для первой и второй производных производящей функции по разным ее компонентам при значении всех переменных компонент вектора u : ui = 1.
Производящая функция может быть записана для каждого из элементов матриц-языков R,M, U. Существует теорема декомпозиции (Regnier, Szpankowski, 1998;
Regnier, 2000), позволяющая представить производящую функцию F ( z, u ) в виде комбинации производящих функций, построенных для элементарных языков R,M, U. Элементарным языкам-матрицам R и M сопоставляются матричные производящие функции R(z,u) (вектор, с компонентами, содержащими вероятности слов, заканчивающихся на разные H j, и не содержащих других вхождений слов и из H ) и M(z,u) (квадратная матрица с ячейками, содержащими все вероятности слов, дополняющих вхождение H i до H j ). Полимодальную производящую функцию ( z, u,..., u ) можно представить в виде разложения производящих FN H,..., N H 1 q 1 q функций элементарных языков R(z), M(z), U(z):
( z, u,..., u ) = Ri ( z ) M ij1 ( z ) M j1 j2 ( z )...M jk 2 jk 1 ( z ) U k 1 ( z ) u1r1...uqq, r FN H,..., N H 1 q 1 q k =1 i, j1,..., jk k, т.е. количество слов типа в наборе ( H1,..., H,..., H q ).
где r = i, + j, = Или в компактной матричной форме: F ( z, u ) = R ( z, u )M k -1 ( z, u ) U* ( z ). В k = главе 4 настоящей диссертации приводятся результаты дифференцирования этой формулы и получены замкнутые аналитические формулы для математического ожидания и дисперсии числа мотивов, встреченных в случайной последовательности, заданной как последовательность независимых случайных испытаний или как марковская цепь первого порядка. В этой главе показано, что в случае независимых случайных испытаний вторая производная для последовательности независимых испытаний имеет вид:
2 F ( z, u ) 2 1 2( H i ( z ) Aij ( z ) + H j ( z ) A ji ( z ) ) ( H ( z ) + H j ( z )) =.
Hi ( z ) H j ( z ) + = (1 z ) i ui u j u =1 (1 z ) (1 z ) = Sind + Soverlap S diag Т.е. в структуре производящей функции выделяются слагаемые соответствующие неперекрывающимся (первое и третье слагаемые) и перекрывающимся (второе слагаемое) словам. Дифференцирование этих слагаемых достаточно прямолинейно.
Для первого слагаемого верно:
2Hi ( z ) H j ( z ) = P ( H i ) P ( H j ) ( n + 2 ) ( mi + m j ) ( n + 1) ( mi + m j ), zn (1 z ) что соответствует суммарной вероятности замостить отрезок длины n неперекрывающимися словами с длинами mi и m j, выраженной через количество таких покрытий. Второе слагаемое более громоздко и имеет вид, включающий все возможные перекрытия слов:
( ) µ 1 µ m l ( ) Soverlap S diag = n P ( H i ) P H j l, m j H i I Hii,H j + P ( H j ) P H i [l, mi ] H j I H jj, Hi ml l =1 l = ( ) µ µ m l ( ) + (1 mi m j ) P ( H i ) P H j l, m j H i I Hii,H j + P ( H j ) P H i [l, mi ] H j I H jj, Hi.
ml l =1 l = ( ) ( ) µ µ + lP ( H i ) P H j l, m j H i I Hii,H j + lP ( H j ) P H i l, m j H j I H jj, Hi m l ml l =1 l = Суммируя различные вклады, из этих формул можно получить выражения для полной дисперсии числа появившихся в последовательности мотивов. Вклад в дисперсию неперекрывающихся появлений мотива равен:
( ) = nP ( H ) 1 P ( H ) 2 P ( H ) + 2P ( H ) P ( H ) + 2P ( H ) i P ( H ) + P ( H ) P ( H ), Vnonoverlap i i i i i i i i i i i i i i P (H ) = P (H ) - полная вероятность появления мотива, а i = mi 1.
где i i В частном случае однобуквенных слов i = 0 эта формула переходит в бернуллиевскую дисперсию V ( H ) = nP ( H ) (1 P ( H ) ). В случае, если все слова мотива имеют одну и ту же длину формула упрощается:
Vnon overlap = nP ( H ) (1 P ( H )(1 + 2 ) ) + ( 3 + 2 ) P 2 ( H ) P ( H ).
К сожалению, для члена с перекрытиями мотивов не удается получить такой же компактной формулы. Однако, в случае, если мотив состоит из единственного слова, формула для дисперсии сводится к уже известной формуле (Regnier, Szpankowski, 1998):
( ) V ( H ) = nP ( H ) ( 2 A (1) ( 2 + 1) ) P ( H ) + P 2 ( H ) ( 3 + 2 ) P ( H ) ( 2 A (1) 1) 2 A (1).
Эти формулы дают точное значение дисперсии числа вхождений мотива в текст, порожденный последовательностью независимых случайных испытаний. На практике часто используется приближение Пуассона, V ~ E.
Для определения применимости этой формулы рассмотрим разность V E, равную:
V E = nP 2 ( H ) ( 2 + 1) + P 2 ( H ) ( 3 + 2 ) + Soverlap ( H i, H j ) S diag ( H i, H j ).
i, j (мы считаем, что мотив содержит слова равной длины). Факторы с ( ) µ перекрывающимися вхождениями типа P ( H i ) P H j l, m j H i I H,H дают при ml i i j l = суммировании вклад порядка nP ( H ). Вклад перекрывающихся мотивов в свободный член, не зависящий от n, также меньше или порядка аналогичного вклада в матожидание, причем величина этого вклада приближается к величине этого слагаемого в матожидание снизу для сильно самоперекрывающихся мотивов (с консенсусами типа AAAAAA).
В случае длинных последовательностей n 1 основную роль играет слагаемое, пропорциональное n. Если вероятность мотива P ( H ) 1, то всеми членами, пропорциональными P 2 ( H ), можно пренебречь, и основной непуассоновский вклад дают перекрывающиеся вхождения мотивов. На практике это означает, что дисперсия меняется только для однородных (типа polyA) или периодических (типа poly-AT) мотивов. Для таких мотивов Пуассоновское приближение неприемлемо. Для остальных мотивов Пуассоновское приближение работает с большой точностью.
В случае, если мотивы не являются маловероятными (при P ( H ) € 1 ), становятся важными поправки, пропорциональные P 2 ( H ). В этом случае пуассоновское приближение очевидно неприменимо, однако Z-значение (см.
ниже) остается хорошим критерием представленности мотива в тексте, в частности, и в случае самопересекающихся мотивов, число появлений которых в последовательности не распределено согласно нормальному закону. При этом, в случае длинных последовательностей достаточно использовать поправки, пропорциональные n. На практике, для конкретного мотива обычно легко оценить вероятность того, что его вхождения будут самоперекрываться. Если число перекрывающихся вхождений невелико, можно ограничиться поправками, для которых не требуется сколько-нибудь сложных вычислений, кроме вычисления вероятности вхождения мотива P (H ).
В случае коротких последовательностей n m следует учитывать члены, не зависящие от n. В случае P ( H ) 1 работает приближение Пуассона, при тех же условиях, что и в случае длинных последовательностей.
Следует отметить, что вычисление дисперсии числа вхождений мотива в последовательность редко является самоценной задачей и обычно используется для построения статистических оценок на перепредставленность или избегание мотива в каких-то выборках последовательностей.
В диссертации также приведено точное вычисление дисперсии для случая, когда случайная последовательность представляет собой цепь Маркова первого порядка. В этом случае появляется дополнительное слагаемое, отражающее корреляцию отдельных слов на близком расстоянии.
Относительный вклад этого слагаемого падает при P ( H ) 1.
Вычисленные матожидание E ( H ) и дисперсия V ( H ) позволяют вычислить P-значение того, что мотив H перепредставлен в данной последовательности, при условии, что E ( H ) 1 и верно нормальное приближение. В этом случае P-значение вычисляется как вероятность «хвоста» нормального распределения, и величиной от которой однозначно O (H ) E (H ), где O ( H ) - это зависит P-значение является Z-значение Z = V (H ) количество реально наблюдаемых вхождений мотива.
Глава 4. АЛГОРИТМИЧЕСКОЕ ВЫЧИСЛЕНИЕ P-ЗНАЧЕНИЯ ДЛЯ ВЕРОЯТНОСТИ ПОСЛЕДОВАТЕЛЬНОСТИ, СОДЕРЖАЩЕЙ МИНИМАЛЬНЫЕ КОЛИЧЕСТВА ВСТРЕЧ КАЖДОГО МОТИВА ИЗ ЗАДАННОГО НАБОРА МОТИВОВ В тех случаях, когда распределение Пуассона неприменимо, для вычисления P-значения приходится использовать алгоритмический подход. В основе подхода лежит модификация алгоритма Ахо-Корасик (Aho, Corasick, 1975). Основная структура данных представляет собой дерево T ( H ), являющееся вариантом бора Trie ( H ) (Knuth, 1975). Дерево строится следующим образом. Пусть Q ( H ) - множество префиксов слов множества H.
Каждый префикс q Q ( H ) отождествляется с узлом Node ( q ). В частности, корень бора отождествляется с пустой строкой. Длина префикса является глубиной узла Node ( q ).
Текст читается слева направо и прочтение каждой буквы сопровождается с передвижением по дереву, задаваемому функцией перехода.
Эта функция для каждого узла и для каждой буквы имеет своими значениями узлы дерева, между которыми осуществляется переход. Значением функции перехода : Q ( H ) Q ( H ) для данной пары {узел, буква} ( p, a ) Q ( H ), является ( p, a ) - наибольший суффикс конкатенации pa, принадлежащий к Q ( H ). Заметим, что ( p, a ) = pa iff pa Q ( H ).
Пусть T [i ] - буква текста T на позиции i. Обозначим как qi самый большой суффикс текста T [1]...T [i ], принадлежащий Q ( H ).
Последовательность узлов, определенная словами qi, строится по индукции i 0, qi +1 = ( qi, T [i + 1]), с начальным условиям q0 =. Такая последовательность называется Ахо-Корасик-обходом бора Trie ( H ), ассоциированного с текстом Tk : {q0,..., qk } = AC (Trie ( H ), Tk ).
Конкретный регуляторный сегмент эукариотической ДНК может содержать достаточно большое число сайтов связывания для различных факторов, причем эти сайты могут перекрываться друг с другом. В результате естественно возникает проблема изучения мотивов, совместно встречающихся в одном и том же сегменте ДНК. Для оценки статистической значимости совместного наблюдения ряда мотивов необходимо вычислить P значения P ( Ln ( H1,..., Hs ;
k1,..., ks ) ) того, что мотивы (H1,..., H ss ) имеют соответственно как минимум ( k1,..., ks ), возможно перекрывающихся, вхождений в текст Tn длины n, записанный в алфавите.
Рисунок 1. Обход дерева Trie ( H ). Мотив задан как множество H = {AAA, AAC, ACA, ACC, CCT}. Синими стрелками показаны переходы, выходящие из вершины ААС, соответствующие каждой возможной следующей букве текста. Красными стрелками показаны переходы, выходящие из вершины СС, соответствующие каждой возможной следующей букве текста.
Пусть дано s различных мотивов (H1,..., Hss ). Упомянутое P-значение будет также обозначаться как P (Tn ( H1,..., H s ;
k1,..., ks ) ). Рассмотрим мотив H = H1 U... U H s являющийся объединением искомых мотивов, т.е. включающий в себя все слова, принадлежащие хотя бы одному из мотивов Hi. Построим бор Trie ( H ) для полного множества слов H. Узел q Q ( H ) может принадлежать как какому-то одному конкретному мотиву Hk либо одновременно некоторому подмножеству мотивов {H j }0 j s. Функция перехода : Q ( H ) Q ( H ) определяется также, как она была определена для случая единственного мотива H.
Все тексты Ti длины i подразделяются на классы, в зависимости от наличия в них вхождения различных мотивов H j. Вводится индекс вхождений I ( k,...,k ) ( l1,..., ls ), вычисляемый для набора мотивов (H1,..., Hss ) и 1 s показывающий, что текст Tn содержит li, возможно перекрывающихся вхождений мотива Hi. Индекс представляет собой s -вектор, i-я координата которого может быть вычислена следующим образом:
l if l ki I ( k,...,k ) ( l1,..., ls ) = i = i i.
1s i ki if li ki Далее определяются классы Ci ( 1,..., s ;
q ), 0 i ki, содержащие тексты Ti длины i, для которых индекс вхождений мотивов (H1,..., H s ) в текст Ti равен AC (Trie ( H ), Ti ) заканчивается в узле q. Кроме того ( 1,..., s ), а обход бора определяются дополняющие классы Gi ( k1,..., ks ).
Для вычисления искомой вероятности производится суммирование по всем узлам дерева q и по всем символам алфавита a. Пусть q - это значение функции перехода ( q, a ) для такой пары (узел, буква). Такое значение может принадлежать сразу нескольким мотивам {H j }0 j s. Тогда, вероятность P ( Ci +1 ( 1,..., s, q ') ) того, что текст Ti принадлежит классу Ci +1 ( 1,..., s ;
q ) может быть вычислена как:
( ) P ( Ci +1 ( 1,..., s, q ' ) ) = P Ci ( I ( r1,..., rs ), q ) * p ( a ), ( q, a ): ( q, a ) = q ', q '{H j }0 js ( li 1) if q Hi где ri =.
li if q Hi Искомая вероятность получается тогда, когда достигается необходимое число вхождений всех мотивов.
Техника, основанная на обходе бора, может быть естественным образом распространена на вычисления P-значения числа вхождений мотива в случайном тексте, порожденном цепью Маркова порядка K (Roytberg, 2007).
Трудоемкость определяется временами порядка ( ) k, где K - это размер алфавита, H - это полное On m H + K i i количество слов в объединенных мотивах, m - это максимальная длина слова, K - порядок цепи Маркова, а ki - минимально допустимое количество вхождений мотива i.
В диссертации рассматриваются также эффективные способы построения дерева-бора в случае, если мотив задан не в виде перечисления слов, а в виде матрицы позиционных весов, с конкретным порогом. В этом случае бор можно построить без явного выписывания всех слов.
Глава 5. РАЗБИЕНИЕ ПОСЛЕДОВАТЕЛЬНОСТИ НА УЧАСТКИ, ОДНОРОДНЫЕ ПО НУКЛЕОТИДНОМУ СОСТАВУ, МЕТОДОМ МАКСИМИЗАЦИИ БАЙЕСОВСКОГО ПРАВДОПОДОБИЯ Иерархическая структура генома, отражающается в иерархии доменов ДНК, имеющих сходный нуклеотидный состав (Frank, Lobry, 1999). Можно предположить, что это связано с различными условиями к термодинамике формирования структур ДНК соответствующих уровней.
В этой главе описывается алгоритм разбиения последовательности нуклеотидов на сегменты, которые могут приближенно считаться статистически однородными. Алгоритм состоит из двух стадий. На первой стадии с помощью динамического программирования находится оптимальная конфигурация границ, разбивающих последовательность на сегменты таким образом, что некоторая функция правдоподобия достигает на этом разбиении своего максимума. На второй стадии алгоритма вычисляется вес каждой найденной границы. Удаление границ с небольшими весами позволяет получить субоптимальную сегментацию, устойчивую к малым вариациям исходной последовательности.
Пусть задан текст T, длины n, записанный в алфавите, состоящем из L = символов. Целью алгоритма является построение оптимального разбиения T на некоторое множество сегментов {T1,..., TK }. Текст T моделируется как реализация некоторой вероятностной модели. В отличие от предыдущих разделов предполагается, что исходный текст представляет собой серию сегментов {T1,..., TK +1}, в каждом сегменте текст порожден как последовательность независимых случайных испытаний и параметры модели в пределах одного сегмента не меняются, причем сегменты предполагаются вероятностно независимыми друг от друга. Сегментацией конкретной последовательности будем называть набор границ {b1,..., bK } разбивающий текст T на сегменты. Каждому сегменту Ti сопоставляется вектор отсчетов – число встреченных букв каждого типа n = (n1,.., n L ).
При поиске оптимальной сегментации каждому сегменту соответствует зависящая от отсчетов весовая функция – мера однородности сегмента.
Полная весовая функция текста, состоящего из ряда сегментов равна сумме весовых функций каждого из сегментов. Для построения меры однородности сегмента используются методы байесовской статистики. Для каждого состава = {1, 2,..., L} задается функция плотности вероятности составов p(), L которая определена на симплексе = { : k 0;
k = 1} и удовлетворяет k = условию нормировки dp( ) = 1. Теорема Байеса позволяет вычислить постериорную плотность распределения вероятностей p( S ) на сегменте () P S p( ), где P(S ) = d P(S ) p( ) это нормировка, называемая S: p( S ) = P (S ) маргинальным правдоподобием, зависящая только от вектора отчетов n и не меняющаяся при перестановках букв в последовательности S. Здесь ( ) i =1 in L P S = – вероятность реализации данной последовательности при известном составе, а ( ) - некоторое априорное распределение на пространстве составов. В задаче сегментации выбор априорного распределения неявно означает определенное предположение о составе полимера, в наших расчетах используется однородное (неинформативное) распределение.
В качестве меры однородости сегмента используется значения его маргинального правдоподобия. Для однородного априорного распределения маргинальное правдоподобие может быть получено аналитически (Liu, Lawrence, 1999):
(L 1)!
P(S ) = P(n ) = n1!...n L !.
( N + L 1)!
Вычисление оптимальной сегментации проводится согласно алгоритму, близкому к алгоритму Витерби (Forney, 1973). Пусть дана последовательность S = s1s2s3… sN длины N, где si. Пусть для каждого сегмента S(a,b) = sa… sb, a b может быть вычислен вес W(a,b). В нашем случае примем этот вес за равный ln ( P ( S ( a, b ) ) ).
Каждая конкретная сегментация R, имеющая m сегментов, определяется набором границ R = {k0 = 0, k1,..., km1, km = N }, где граница ki стоит между символами sk и sk. Определим вес всей сегментации R i + i ( ) m F ( R ) = W k j 1 + 1, k j. Для вычисления оптимальной сегментации R, которая * j = максимизирует функционал F ( R ), используется рекуррентный алгоритм, описанный в (Roytberg, Finkelstein, 1993). Обозначим R* ( k ) оптимальную сегментацию фрагмента последовательности S (1, k ), 1 k N. Сегментация R* (1) – тривиальна. В случае, если известны оптимальные сегментации всех участков R* (1),..., R* ( k 1), можно найти оптимальную сегментацию участка [ ] R* ( k ) используя рекуррентное выражение: F ( R * ( k )) = max F ( R * (i )) + W (i + 1, k ), i = 0,..., k c начальным условием F(R*(0)) = 0.
Более стабильной характеристикой является статсумма на множестве сегментаций последовательности. Статсумма вычисляется как сумма весов всевозможных сегментаций Z (N ) =... (q1,... qN 1 ), где индикатор qk равен q1 qN единице, при границе после позиции k и нулю в других случаях, а сегментация, заданная вектором границ q = ( q1,...qN ), имеет вес (вероятность) (q).
Статистическая сумма может использоваться для оценки вклада наличия границы на позиции k в разнообразные сегментации. Для этого необходимо вычислить статистические суммы сегментаций фрагментов последовательности, ZL(k) и ZR(k) находящихся справа и слева от данной границы. Для вычисления этих значений могут использоваться следующие k 1 N ZL( j ), ZR( k ) = eW ( k, j ) ZR( j ) с реккурентные формулы: ZL( k ) = e W ( j + 1, k 1) j=0 j=k соответствующими граничными условиям.
Статистический вес границы, расположенной после позиции k, может Z L ( k )Z R (N k ) быть вычислен как ( qk = 1) =. Границы с более высоким Z (N ) статистическим весом разделяют сегменты, которые сильнее различаются по своему составу.
Глава 6. ВЫДЕЛЕНИЕ ХАРАКТЕРНЫХ МОТИВОВ ИЗ ВЫБОРОК НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ С ПОМОЩЬЮ АЛГОРИТМА ГИББСОВЫХ ВЫБОРОК В главах 3 и 4 было рассмотрено вычисление статистической значимости встречаемости мотивов в ДНК. Экспериментальные данные, такие как SELEX и футпринтинг с ДНКазой I, позволяют получить мотивы для каждого регуляторного белка;
для этого используются методы определения мотивов (motif discovery). В данной главе предлагается новый метод определения мотивов, учитывающий симметрию участков ДНК, непосредственно взаимодействующих с регуляторным белком. Исходной точкой послужил алгоритм Gibbs sampling (Lawrence, 1993).
В алгоритме SeSiMCMC (сокращенно от «Sequence Similarity Markov Chain Monte Carlo) сохранено представление мотива в виде МПВ. Каждая исходная последовательность сегментируется на «мотив» и «фон», порожденные разными вероятностными моделями. Фон моделируется как однородная последовательность независимых случайных испытаний.
Критерием оптимальности сегментации считается ее максимальная вероятность, вычисленная в рамках Байесовского подхода, при условии известной последовательности (ДНК данных) и отсутствия априорной информации о составе ДНК и предпочтения определенных нуклеотидов в каждой позиции мотива. Выровненная часть набора последовательностей определяет мотив. Оставшиеся невыровненными части всех последовательностей считаются фоном.
Для выровненной части, для каждой позиции выравнивания можно вычислить позиционные числа встречаемости нуклеотидов ni,, из которых можно оценить позиционные вероятности qi, появления нуклеотида в ni, + p позиции i, i = 1..m, где m - это длина выравнивания, как q ( i, ) =. При M +B g + b этом для фонового распределения принимается оценка f ( ) =. Здесь M K+B - это число выровненных последовательностей, K - это число всех невыровненных (фоновых) позиций во всех входных данных, а g - полное число нуклеотидов типа в невыровненных позициях. Псевдокаунты b выбираются пропорциональными частотам нуклеотидов во входных данных, в то время как их сумма B = b ~ N, где N - это число входных последовательностей.
Если принимается, что мотив имеет структуру прямого повтора, то ni, + n + 2 b m +, i +int МПВ оценивается по формуле q ( i, ) =, в то время как для 2 (M + B) палиндромов (обратно-комплементарных повторов) формула принимает ni,r + nm+1i, + b + b q ( i, ) = 2 (M + B), где m - длина мотива и — это нуклеотид, вид:
комплементарный.
Алгоритм действует следующим образом. Вначале формируется выравнивание, состоящее из случайно выбранных участков некоторой длины, по одному на последовательность. Исходная длина либо выбирается случайно в некоторых пределах, либо задается как параметр программы. Затем заданные последовательности по очереди просматриваются (в цикле). На каждом шаге выбирается «текущая» последовательность. Далее, по выравниванию, включающему в себя все последовательности, кроме «текущей», строится МПВ. Сегменты последовательностей, не вошедшие в выравнивание, считаются порожденными из фонового распределения. После того, как оценены МПВ и фоновое распределение, вычисляется вероятность получить текущую последовательность, при условии, что мотив расположен в позиции k, и для каждой его позиции принята модель, взятая из соответствующей колоноки МПВ:
k 1 k + m1 L m+ P (T | [ k ], q, f ) = f ( ri ) q ( i k + 1, ri ) f ( r ), k 0;
i i =1 i =k i=k +m L m+ P ( R | [ 0]) = f ( r ),i i = здесь ri — это i -ый нуклеотид в последовательности T, а [ k ], k = 1..( L m + 1) обозначают событие: «сайт начинается с позиции k », [0] соответствует случаю отсутствия сайта (нулевая позиция). Априорная вероятность P ([0]) определяется пользователем и обозначает вероятность того, что последовательность из входных данных является шумом и не несет никакой биологической информации. Все ненулевые позиции имеют равные априорные вероятности.
Апостериорная вероятность того, что сайт начинается в позиции k P (T | [ k ], q, f ) P ([ k ]) P ( T | [ k ], q, f ) P ( [ k ] ) равна P ([ k ] | R, q, f ) =. Из этого = P ( T | q, f ) P ( T | q, f ) апостериорного распределения разыгрывается новая (возможно нулевая) позиция сайта. После этого полученная позиция сайта считается заданной для текущей последовательности во всех последующих шагах цикла. Далее выбирается следующая текущая последовательность, и цикл повторяется.
Процесс последовательных итераций продолжается до тех пор, пока цепь Анализ Чтение и Создание и аргументов и верификация инициализация конфигурацио входных внутренних нного файла данных структур Выбор типа Увеличение Оценка следующего шага счётчика числа предполагаемого изменения шагов, его изменения качества набора паттернов паттернов, сравнение с розыгрыш нового заданным паттерна числом шагов Решение: изменение принимается или не Новое состояние принимается, во заносится в втором случае новое статистику состояние повторяет старое Анализ собранной за Вывод результатов время работы статистики Рисунок 2. Блок-схема работы алгоритма SeSiMCMC.
полученных МЛАБП не сойдется (то есть, изменения от шага к шагу не станут малыми). Мотив определяется из полного набора текущих положений сайтов, который на рис.2 называется «паттерном».
Кроме местоположения мотива определяется оптимальная длина мотива и длины промежутка, если разрешены мотивы, состоящие из двух частей. Для каждой длины мотива длина спейсера принимается как минимальное значение, для которого достигается локальный максимум ИСП. Поскольку длина спейсера может оказаться нулевой, эта же процедура определяет, разделен мотив или нет. Блок-схема программы приведена на рис. 2. Для оптимизации длины мотива и спейсера может использоваться один из двух протоколов. Используемый по умолчанию быстрый режим выполняет оптимизацию на каждом уточнении локального выравнивания, как описано выше. В медленном режиме длина мотива ступенчато изменяется от наименьших значений к наибольшим и полная процедура поиска мотива без оптимизации длины выполняется для каждой ступени.
Глава 7. ВЫЯВЛЕНИЕ ЦИС-РЕГУЛЯТОРНЫХ МОДУЛЕЙ КАК СЕГМЕНТОВ ДНК СОДЕРЖАЩИХ КЛАСТЕРЫ СХОДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ, ОБЕСПЕЧИВАЮЩИХ КООПЕРАТИВНЫЕ ДНК БЕЛКОВЫЕ ВЗАИМОДЕЙСТВИЯ В этой главе диссертации приведены полученные с помощью всего арсенала разработанных методов результаты идентификации в цис-регуляторных модулях кластеров сайтов связывания ССТФ белков, регулирующих систему генов, ответственных за раннее развитие Drosophila melanogaster. В результаты включены данные ручной переаннотации большого количества экспериметальных данных, относящихся к ЦРМ (энхансерам), которые управляют ранним эмбриональным развитием Drosophila. Все данные, полученные в ходе выполнения этой работы, включены в межународную базу данных RedFly (Halfon, 2008) известных генетических элементов Drosophila melanogaster. В результате работы получена информация трех типов: (1) сайты связывания транскрипционных факторов (ССТФ) для известных регуляторных белков;
(2) известные ЦРМ в ДНК и (3) информация о зависимости конкретных генов, входящих в систему, от конкретных факторов (регуляторные взаимодействия).
Факторы, регулирующие развитие Drosophila, и распознаваемые ими мотивы ДНК. Для анализа мотивов использованы 28 регуляторных белков, участвующих в регуляции ранних стадий развития Drosophila, включая материнские гены, гап-гены, гены pair rule, и ряд генов, управляющих полярностью сегментов. Мы использовали экспериментальные данные о связывании этих белков с ДНК полученные in vitro, преимующественно футпринтами с ДНКазой I и SELEX. Использованы также данные о специфических мутациях и эволюционной сохранности соответствующих участков ДНК.
Матрицы позиционных весов (МПВ) были построены с помощью программы SeSiMCMC (см. гл. 6). Подробное описание данных находится на сайте http://line.imb.ac.ru/DMMPMM/. Наилучшие мотивы для нескольких факторов приведены в табл. 1, относительная высота букв отражает вклады соответствующих букв в информационное содержание данной позиции.
Bcd Cad Gt Hb Kni Kr Таблица 1. Некоторые мотивы, распознаваемые регуляторными белками, рассматривавшимися в исследовании. Дополнительная информация содержится на сайте http://line.imb.ac.ru/DMMPMM/ Исследованы гены: btd, gt, hb, kni, kr, otd, sal, tll, eve, ftz, gsb, h, run, en, dll, ems, ubx и slp, управляющие ранним развитием Drosophila, в первую очередь, управляющие осевой дифферецировкой яйца мухи. Длины их локусов варьируют между 16 и 120 тыс. п.о. В локусах картированы цис регуляторные модули, известные из литературы, всего около 60 ЦРМ.
.
Рисунок 3. Локус гена even-skipped с картированными модулями: (CDS) кодирующая область;
(P) проксимальный промотор;
(late1) поздний энхансер late1;
(3+7) энхансер eve stripe 3+7;
(2) энхансер eve stripe 2;
(late2) поздний энхансер late2;
(1) энхансер eve stripe 1;
(5) энхансер eve stripe 5.
Анализ распределения мотивов связывания регуляторных факторов в ДНК проводился путем сканирования последовательности ДНК с помощью МПВ, построенных для различных факторов, и подсчета количества найденных сайтов в скользящем окне фиксированной длины. Важными параметрами служили пороги для МПВ для каждого из белков.
Гомотипические кластеры мотивов связывания белка Bicoid в локусе гена even-skipped. Простейшим вариантом конфигурации сайтов являются гомотипические кластеры, т.е. участки повышенной плотности ССТФ для одного и того же белка. Для определения кластеров подсчитывается число ССТФ, найденных в скользящем окне. На рис. показаны кластеры участков связывания белка Bicoid в локусе гена even skipped, полученные для разных значений длины окна и порога МПВ. На панели (А) рисунка приведена зависимость статистической значимости полученых кластеров от порога МПВ. На панели (B) показана зависимость статистической значимости как функция размера сканирующего окна. Во всех случаях порог МПВ выбран равным 5,5, что показано красной стрелкой на оси Y на панели (А). При этом кластер содержит пять сайтов, причем вероятность появления каждого из сайтов приблизительно оценивается в 1 на 1000 п.о.
Видно ступенчатое поведение зависимости статистической значимости кластера от размера окна. При больших размерах окна, в него может попасть большее число сайтов связывания, однако, статистическая значимость кластеров, содержащих одинаковое число сайтов связывания, уменьшается с ростом размера окна. На рис. 4B видно также сложное строение кластеров связывания белка Bicoid. В частности, видно, что при малых размерах окна кластер, соответствующий энхансеру eve stripe 1 распадается на два подкластера из которых правый подкластер, возможно, относится уже к энхансеру eve stripe 5. Замечательно, что из семи энхансеров гена eve только три (stripe 2, stripe 1, и сливающийся с последним stripe 5) участвуют в регуляции с помощью белка Bicoid. Именно эти энхансеры и содержат кластеры сайтов связывания белка Bicoid, как это видно из рис. 4.
При этом кластер в энхансере stripe 5 смещен влево и сливается с кластером в энхансере eve stripe 1. Таким образом, анализ гомотипических кластеров регуляторных факторов в последовательности ДНК может предсказать как местоположение энхансеров в пределах локуса, так и зависимость этих энхансеров от определенных регуляторных факторов.
Однако при таких предсказаниях возможны артефакты. На рис. присутствует также кластер сайтов связывания, расположенный в пределах энхансера late2, по-видимому, имеющий отношение ССТФ белка Paired (Prd), мотив которого достаточно похож на мотив связывания Bcd.
Оптимизация параметров поиска гомотипических кластеров мотивов связывания факторов регуляции транскрипции в локусах генов раннего развития Drosophila. Как видно из рис. 4 гомотипические кластеры мотивов, распознаваемых белком Bicoid хорошо коррелируют с энахансерами гена even-skipped, зависимыми от этого транскрипционного фактора.
Степень скореллированности различна при разных порогах МПВ и разных размерах сканирующего окна. Для предсказания местоположения новых энхансеров имеет смысл определить оптимальное значение порога МПВ и оптимальную длину окна, при которых наибольшее количество известных энхансеров определяется достаточно точно, и в то же время ошибка перепредсказания достаточно мала. В общей сложности белок Bicoid регулирует восемь генов: tll, otd, btd, sal, hb, kr, kni, eve. Нуклеотидные последовательности локусов всех этих генов были просканированы МПВ Bicoid, после чего на разных уровнях порога МПВ были получены кластеры сайтов путем сканирования окнами различной длины.
Рисунок 4. Распределение кластеров мотивов распознаваемых фактором Bicoid в локусе гена even-skipped. Справо вверху: цветовой код статистической значимости (отрицательный логарифм вероятности возникновения кластера) показан цветом. Ось X - позиция в локусе.
Внизу: карта локуса even-skipped (см. Рис. 3). Ось Y: (A). Переменный порог МПВ при фиксированном скользящем окне 500 п.о. (B). Переменный размер скользящего окна, при фиксированном пороге МПВ 5.50.
Для каждого положения окна, каждой длины окна, и каждого порога МПВ вычислялась вероятность найти наблюдаемое количество сайтов связывания в случайной последовательности по формуле Пуассона с поправками, полученными в глааве 3. Вероятность p появления индивидуального сайта с весом, большим избранного порога, определялась эмпирически, путем оценки частоты сайтов в полном геноме Drosophila, при условии удаления из генома мобильных элементов и микро- и минисателлитов большой длины. Был введен порог на вероятности получения гомотипических кластеров взаимодействующих мотивов P ( S ( HT, r, li ) ). Все кластеры, имеющие веса, превышающие этот порог, считались «предсказанием» для данной длины окна и данного порога МПВ. Эти предсказанные кластеры сравнивались с экспериментально определенными энхансерами в восьми генах, зависящих от Bcd. Для оценки качества сходства (степени перекрывания вычисленных гомотипических кластеров мотивов и экспериментально определенных ЦРМ) использовался коэффициент ассоциации Пирсона СС (Mathews, 1998). Выбирались такие размеры окна, порог веса, вычисленного с помощью МПВ и порог на вероятность кластера P ( S ( HT, r, li ) ), для которых значение CC было максимальным. Оказалось, что максимум достигается при пороге МПВ, равном 4,2, длине окна 550, и пороге на вероятность кластера, равном 4*10-4. При этих соотношениях глобальное значение коэффициэнта ассоциации CC оказывается равным 0,62, что является очень хорошим результатом, поскольку границы многих энхансеров определены достаточно неточно, методом грубого делеционного анализа, с рассмотрением небольшого числа делетированных участков разной длины, содержащих энхансер.
Гомотипические кластеры мотивов связывания различных регуляторных факторов в локусах, управляющих ранним развитием Drosophila. Для того, чтобы выяснить насколько отмеченный эффект характерен для генов, регулирующих раннее развитие Drosophila, был предпринят поиск гомотипических кластеров мотивов, распознаваемых более чем 16 факторами, в локусах 20 генов Drosophila, содержащих более энхансеров. Мотивы для 12 факторов оказались недостаточно надежно установленными и полученными по малому количеству данных, а число ЦРМ, зависящих от этих факторов, оказалось недостаточно велико, чтобы надежно определить параметры поиска. Значения длины сканирующего окна, порога МПВ и порога кластера подбирались подобно тому, как это было описано в предыдущей секции для кластеров Bicoid. Для кажой пары (мотив, локус) длина сканирующего окна и порог МПВ были фиксированы, однако порог на статистическую значимость кластера в некоторых случаях зависел от конкретного локуса и мотива. После оптимизации по 60 регуляторным областям были получены оптимальные значения, приведенные в Таблице 2.
Видно, что окно длиной 575 приводит к удовлетворительным результатом для девяти мотивов. Кроме того, положительным результатом является то, что оптимальный порог на МПВ для всех факторов соответствует вероятности появления приблизительно одного вхождения мотива на тысячу пар оснований. При использовании этих оценок длины окна и порога по МПВ оказалось возможным просканировать все имеющиеся локусы МПВ для всех имеющихся факторов и построить приблизительную сеть генетической регуляции. Результаты сканирования всех имеющихся локусов приведены в таблице 2. Как видно из таблицы 2 гомотипические кластеры мотивов связывания позволяют предсказать регуляторные модули далеко не для всех факторов. Если для материнских градиентов и для gap-генов получается достаточно хорошее предскание, то предсказание для генов paired-rule уже значительно уступает по качеству.
Любопытно, что даже в случае факторов Bcd и Cad, для которых получаются весьма удовлетворительные предсказания, существуют локусы, в которых ЦРМ предсказываются весьма плохо. Так, из таблицы 2 видно, что для большинства локусов, зависимых от Bcd, для которых средние характерные значения корреляций имеют порядок 0,6-0,7, получаются очень плохие предсказания в генах otd и tll. Аналогично, для Cad наблюдается блестящая корреляция в локусах gt и tll, но отсутствует корреляция в локусах sal, en и ftz. Следует отметить, что регуляция otd посредством Bcd была показано экспериментально (Ochoa-Espinosa, 2005), так что в этом случае приходится говорить о истинном недопредсказании метода.
Использование программы кластеризации для предсказания регуляторных элементов в геномах и анализа их структуры Набор програмных скриптов CLUSTER (Lifanov, 2003), в котором реализован алгоритм, описанный выше, использовался рядом исследователей для предсказания положения регуляторных модулей в геномах и анализа их структуры. В частности, с помощью описываемой программы был предсказан ряд регуляторных модулей, в частности модуль, регулируемый Bcd в гене gt, что в дальнейшем подтвердилось в экспериментальном исследовании. Полное сканирование генома показало, что в полном геноме существует 14 сильных кластеров Bcd из которых для 11 была показала Bcd-зависимая регуляторная активность (Ochoa-Espinosa, 2006).
Таким образом использование кластеризации мотивов связывания регуляторных белков позволило удвоить число известных ЦРМ, зависящих от Bcd. В таблице 3, взятой из этой работы приводятся точные координаты новых энхансеров, зависящих от Bcd, найденных с помощью нашей программы кластеризации мотивов.
Другое возможное приложение описанного алгоритма – анализ структуры кластеров мотивов, присутствующих в различных регуляторных модулях. Очевидно, что одно и то же значение статистической значимости кластера можно обеспечить различными способами. Во-первых, кластер может включать в себя небольшое количество сильных сайтов связывания регуляторного белка, во-вторых, кластер может содержать большое число сайтов, с небольшими значениями веса, расчитанного с помощью МПВ.
Рисунок 5. Поиск максимума корреляции между положением кластеров мотивов связывания Bicoid и положеним зависимых от Bicoid ЦРМ в последовательностях восьми генов. Цветом показано значение коэффициента ассоциации Пирсона (СС). Ось X: значения порога по МПВ.
Ось Y: размер сканирующего окна. Панели – порог по статистической значимости кластеров: (А) P=0.0005;
(B) P=0.001;
(С) P=0.005. Максимальное значение достигается при пороге МПВ 4.2, длине окна 550 п.о., и пороге статистической значимости кластера 0.0005.
В работе (Berg, von Hippel, 1982) было показано, что вес сайта по МПВ коррелирует с аффинностью этого участка ДНК к соответствующему регуляторному белку, а кластер ССТФ выполняет свою функцию при условии посадки молекул регуляторного белка на достаточно большое количество связывающих ССТФ. Таким образом наличие гомотипического кластера сайтов обеспечивает большую суммарную аффинность к кооперативно взаимодействующим регуляторным белкам.
В этом случае энхансер, содержащий небольшое число сильных сайтов, будет работать как при высоких, так и при низких концентрациях регуляторного белка. В то же время, энхансер, содержащий большое число слабых сайтов будет работать только при высоких концентрация регуляторного белка и не будет работать при его низких концентрациях.
Таким образом структура кластера ССТФ может быть использована для анализа фунций конкретного регуляторного элемента.
Такой анализ был проведен, и, действительно, подтвердился экспериментально (Clyde et al. 2005). В этой работе, в частности было показано, что четыре полосы экспрессии гена even-skipped управляются всего двумя энхансерами, реагирующими на встречные градиенты двух регуляторных белков Hb, и Kni (белок Kni экспрессируется в средней части эмбриона, а белок Hb на полюсах). Энхансер eve3+7 содержит слабые сайты связывания Hb и сильные сайты связывания Kni. Поскольку оба белка являются репрессорами, под управлением этого энхансера ген even-skipped экспрессируется ближе к полюсам, в области, где концентрация Hb заметна, но не достаточно велика, чтобы вызвать ответ кластера слабых сайтов. В то же время сильные сайты в кластере мотивов связывания Kni не позволяют энхансеру eve3+7 запускать экспрессию even-skipped в области хоть сколько нибудь заметных концентраций Kni в районе центральной области эмбриона.
Обратная картина наблюдается в случае запуска гена even-skipped в экспрессию благодаря взаимодействию с энхансером eve4+6. Этот энхансер содержит сильные сайты связывания Hb и слабые сайты связывания Kni.
Поэтому, под управлением этого энхансера ген even-skipped запускается в экспрессию в области приближающейся к средней области эмбриона.
Поскольку таким образом образуется по две полосы с переднего и с заднего конца эмбриона, оказывается, что градиентов концентрации всего двух белков достаточно для того, чтобы определить восемь границ четырех полос экспрессии even-skipped! В работе (Clyde 2005) приведены экспериментальные данные по генной инженерии сайтов в энхансерах eve4+ и eve3+7. Изменение силы сайтов этих энхансерах приводило к смещению положения полос экспрессии гена even-skipped.
Ген и ЦРМ Длина Местоположение hb P2 243 0 kb 5' Kr CD1 730 3 kb 5' kni 1,019 1.2 kb 5' tll 1,036 1.5 kb 3' gt1 787 6 kb 5' gt23 1,213 10 kb 5' btd 1,080 3 kb 5' otd early 925 3.3 kb 5' salBE 421 10 kb 5' bowl 388 2nd intron hairy0 470 8 kb 3' hairy2 1,080 8.5 kb 5' hairy7 932 9.5 kb 5' eve1 788 5.5 kb 5' eve2 488 1 kb 5' slpA 372 1 kb 5' of slp slpB 793 2 kb 3' of slp 7 kb 3' of slp prd 1,400 1 kb 3' D/fsh 748 2 kb 3' Таблица 3. Предсказание местоположения энахансеров (ЦРМ), зависимых от Bcd в окресностях различных генов с помощью метода, разработанного в диссертации.
Приведенены экспериментальные данные, полученные в работе [Ochoa-Espinosa et al.
PNAS, 102, 4960], совпадающие с предсказаниями Рисунок 6 Регуляция экспрессии гена eve с помощью градиентов Hb и Kni. Внизу: карта локуса гена eve. (А). Распределение градиентов Hb и Kni и паттерн экспрессии eve.
Экспрессия eve в пределах полос, соединненных скобкой eve3+7 и eve4+6 управляется соответствующими энхансерами. (B). Кластеры мотивов, распознающих Hb. Виден сильный кластер в районе энхансера, управляющего «внутренними» полосами 4 и 6, и более слабый кластер более слабых сайтов в энхансере, управляющем внешними полосами 3 и 7. (С) Тоже для Kni. Сильный кластер присутствует только в энхансере, отвечающем за экспрессию в полосах 3 и 7.
Фактор BCD CAD KR HG ABDB KNI GT PRD EN TLL TTK FTZ Порог МПВ 4.2 4.8 4 6.5 5.4 3.6 7 7.5 4 4.3 3.6 4. 4 3 3 3 3 2 2 2 4 4 * 10 1.2 * 10 2.3 * 10 4.4 * 10 2.3 * 10 1.1 * 10 1.1 * 10 4.1 * 10 3 * 10 1.4 * 10 4.4 * 10^3 6 * Порог Pкл Локус abda 0.05 0. btd 0.918 0.07 0. dll ems 0.771 en 0 0. eve 0.547 0.459 0.246 0.413 0.544 0. ftz 0 0.493 0. gsb 0. gt 0.93 0.754 0. hairy 0.779 0.376 0.476 0. hb 0.417 0.758 0. kni 0.62 0.404 0.933 0. kr 0.702 0.06 0.615 0. otd 0. Таблица 4 Сравнение положения гомотипических кластеров мотивов связывания регуляторных белков и экспериментально определенных ЦРМ для различных энхансеров. Пустые клетки соответствуют генам, для которых не показана зависимость от конкретного фактора.
Выводы 1. Установлены специфические особенности последовательностей ДНК, определяющие архитектуру взаимодействия ДНК с белковым фактором и обеспечивающие иерархическую организацию компонент генома.
2. Оценена вероятность разномасштабных флуктуаций первичной структуры в модельной случайной последовательности ДНК, в результате которых самопроизвольно возникают участки специфического связывания регуляторных белков.
3. Проведена сегментация последовательности генома на разных масштабах на участки, однородные по своему нуклеотидному составу.
Сегментация осуществлялась с помощью вычисления статсуммы всевозможных разбиений последовательности на сегменты, что позволило выявить участки ДНК, которые могут быть описаны статистическими моделями.
4. Локализованы участки ДНК, соответствующие специфическому взаимодействию с регуляторными белками-факторами. Большая точность локализации достигнута за счет учета симметрии структуры ДНК-белок, при взаимодействии с регуляторным фактором в форме димера.
5. Проведена идентификация регуляторных сегментов ДНК (промоторы и энхансеры) как участков ДНК, имеющих высокую афинность к кооперативно связывающимся белковым факторам.
6. Показано, что кооперативность взаимодействия регуляторных факторов с ДНК позволяет сформировать сложную картину экспрессии генов в пространстве развивающейся личинки Drosophila, под управлением трехмерных градиентов небольшого числа регуляторных белков.
Публикации по теме работы Полищук М.С., Хайнцель, А., Фаворов А.В., Макеев В.Ю.
Сравнительный анализ участков связывания белков-регуляторов транскрипции в раннем развитии Drosophila Melanogaster, определенных методом ChIP-chip, и вычислительно предсказанных кластеров сайтов связывания этих белков. Биофизика. 2008, 53(5):754- Bogush VG, Sokolova OS, Davydova LI, Klinov DV, Sidoruk KV, Esipova NG, Neretina TV, Orchanskyi IA, Makeev VY, Tumanyan VG, Shaitan KV, Debabov VG, Kirpichnikov MP. A novel model system for design of biomaterials based on recombinant analogs of spider silk proteins. J Neuroimmune Pharmacol.
2009 Mar;
4(1):17-27.
Лифанов А.П., Власов П.К., Макеев В.Ю., Есипова Н.Г. Нуклеосомный повтор и расположение экзонов и интронов в генах коллагенов типов I и VII Биофизика. 2008, 53(3):524-8.
Рахманов С.В., Макеев В.Ю. Использование невзаимодействующих проб в пространстве белковой структуры для построения статистических потенциалов межатомного взаимодействия Биофизика, 2008;
53(3):389-96.
Britanova LV, Makeev VJ, Kuprash DV. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem Biophys Res Commun. 2008 Jan 18;
365(3):583-8.
Boeva V, Clment J, Rgnier M, Roytberg MA, Makeev VJ. Exact p-value calculation for heterotypic clusters of regulatory motifs and its application in computational annotation of cis-regulatory modules. Algorithms Mol Biol. Oct 10;
2:13.
Enikeeva FN, Kotelnikova EA, Gelfand MS, Makeev VJ. A model of evolution with constant selective pressure for regulatory DNA sites. BMC Evol Biol. 2007 Jul27;
7:125.
Rakhmanov SV, Makeev VJ. Atomic hydration potentials using a Monte Carlo Reference State (MCRS) for protein solvation modeling. BMC Struct Biol.
2007 Mar 30;
7:19.
В.А. Боева, М.В. Фридман, В.Ю. Макеев Эволюция микро- и мнинисателлитов в геноме человека. Биофизика. 2006, 51:650-655.
Е.Д.Ставровская, В.Ю.Макеев, А.А.Миронов CLUSTERTREE-RS:
алгоритм кластеризации регуляторных сигналов с помощью бинарного дерева. Молекулярная биология. 2006, 40: 524-532.
Malko DB, Makeev VJ, Mironov AA, Gelfand MS. Evolution of exon-intron structure and alternative splicing in fruit flies and malarial mosquito genomes.
Genome Res. 2006 Apr;
16(4):505-9. Epub 2006.
Boeva V, Regnier M, Papatsenko D, Makeev V. Short fuzzy tandem repeats in genomic sequences, identification, and possible role in regulation of gene expression. Bioinformatics. 2006 Mar 15;
22(6):676-84.
Favorov AV, Gelfand MS, Gerasimova AV, Ravcheev DA, Mironov AA, Makeev VJ. A Gibbs sampler for identification of symmetrically structured, spaced DNA motifs with improved estimation of the signal length. Bioinformatics. May 15;
21(10):2240-5..
Kotelnikova EA, Makeev VJ, Gelfand MS. Evolution of transcription factor DNA binding sites. Gene. 2005 Mar 14;
347(2):255-63.
Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Rgnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005 Jan;
23(1):137 44.
Рагулина, Л.Е., Макеев, В.Ю., Есипова, Н.Г., Туманян, В.Г., Богуш, В.Г., Дебабов В.Г. Анализ вторичных структур спидроинов первого и второго типов из пауков, принадлежащих различным видам. Биофизика, 2004;
49(6):1147-9.
Рагулина, Л.Е., Макеев, В.Ю., Есипова, Н.Г., Туманян, В.Г., Никитин, А.М., Богуш, В.Г., Дебабов В.Г.Исследование периодичностей в последовательностях аминокислот спидроинов первого и второго типов из пауков различных видов. Биофизика, 2004, 49(6) 1053- Kattenhorn LM, Mills R, Wagner M, Lomsadze A, Makeev V, Borodovsky M, Ploegh HL, Kessler BM. Identification of proteins associated with murine cytomegalovirus virions. J Virol. 2004 Oct;
78(20):11187-97.
Makeev VJ, Lifanov AP, Nazina AG, Papatsenko DA. Distance preferences in the arrangement of binding motifs and hierarchical levels in organization of transcription regulatory information. Nucleic Acids Res. 2003 Oct 15;
31(20):6016 26.
Kalinina OV, Makeev VJ, Sutormin RA, Gelfand MS, Rakhmaninova AB.
The channel in transporters is formed by residues that are rare in transmembrane helices. In Silico Biol. 2003;
3(1-2):197-204.
Vandenbogaert M, Makeev V. Analysis of bacterial RM-systems through genome-scale analysis and related taxonomy issues. In Silico Biol. 2003;
3(1 2):127-43. Epub 2003.
Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. Homotypic regulatory clusters in Drosophila. Genome Res. 2003 Apr;
13(4):579-88.
Кравацкая, Г.И., Франк, Г.К., Макеев, В.Ю., Есипова Н.Г. Сходство периодических структур в расположении нуклеотидов на участках начала репликации бактериальных геномов. Биофизика. 2002. 47(4):595-9.
Papatsenko DA, Makeev VJ, Lifanov AP, Rgnier M, Nazina AG, Desplan C.Extraction of functional binding sites from unique regulatory regions: the Drosophila early developmental enhancers. Genome Res. 2002 Mar;
12(3):470-81.
Ramensky VE, Makeev VJ, Roytberg MA, Tumanyan VG. Segmentation of long genomic sequences into domains with homogeneous composition with BASIO software. Bioinformatics. 2001 Nov;
17(11):1065-6..
Ramensky VE, Makeev VJu, Roytberg MA, Tumanyan VG. DNA segmentation through the Bayesian approach. J Comput Biol. 2000 Feb-Apr;
7(1 2):215-31.
Есипова Н.Г., Кутузова Г.И., Макеев В.Ю., Франк Г.К., Баландина А.В., Камашев Д.Э., Карпов В.Л. Анализ особенностей распределения нуклеотидов на участке начала репликации хромосомы oriC из E. coli. Биофизика, 2000, т.
45, № 3, с. 432-438.
Кривенцева Е.В., Макеев В.Ю., Гельфанд М.С. Статистический анализ экзон-интронной структуры генов высших эукариот. Биофизика, т. 44, № 4, 1999, с. 595-600.
Кутузова, Г.И., Франк, Г.К., Есипова, Н.Г., Макеев, В.Ю., Полозов Р.В.
Периодичности в контактах РНК-полимеразы с промоторами. Биофизика, 1999 Mar-Apr;
44(2):216-23.
Frank GK, Makeev VJ. G and T nucleotide contents show specie-invariant negative correlation for all three codon positions. J Biomol Struct Dyn. Apr;
14(5):629-39.
Кутузова Г.И., Франк Г.К., Макеев В.Ю., Есипова Н.Г., Полозов Р.В.
Фурье-анализ нукле-отидных последовательностей. Периодичности в промоторных последовательностях Есoli. Биофизика, 1997, 42(2):354-62..
Makeev V.Ju, Tumanyan VG. Search of periodicities in primary structure of biopolymers: a general Fourier approach. Comput Appl Biosci. 1996 Feb;
12(1):49 54.
Макеев В.Ю., Франк Г.К., Туманян В.Г. Статистика периодических закономерностей в последовательностях интронов человека М., Наука.
Биофизика, том 41, вып. 1., 1996.
Makeev V.Ju, Tumanyan VG, Esipova NG. The third nucleotide of the Gly coding triplet remembers the periodicity of the collagen chain. FEBS Lett. 1995;
366(1):33-6.
Макеев В.Ю., Туманян В.Г. О связи методов автокорреляционной функции и дискретного анализа Фурье при анализе биологических последовательностей. Биофизика, 1994, Макеев В.Ю. Стохастический резонанс и его возможная роль в живой природе. Биофизика, 1993, 38, 1, ст. 194.